Key Points
Platelet-monocyte interaction engages a reciprocal activation loop that feeds thromboinflammation in COVID-19.
Platelet adhesion is a primary signaling mechanism for monocyte activation that is amplified by tissue factor-dependent signaling.

Abstract
Accumulating evidence into the pathogenesis of COVID-19 highlights a hypercoagulability state with high risk of life-threatening thromboembolic complications. However, the mechanisms of hypercoagulability and their link to hyperinflammation remain poorly understood. Here, we investigate functions and mechanisms of platelet activation and platelet-monocyte interactions in inflammatory amplification during SARS-CoV-2 infection. We used a combination of immunophenotyping, single-cell analysis, functional assays, and pharmacological approaches to gain insights on mechanisms. Critically ill patients with COVID-19 exhibited increased platelet-monocyte aggregates formation. We identified a subset of inflammatory monocytes presenting high CD16 and low HLA-DR expression as the subset mainly interacting with platelets during severe COVID-19. Single-cell RNA-sequencing analysis indicated enhanced fibrinogen receptor Mac-1 in monocytes from patients with severe COVID-19. Monocytes from patients with severe COVID-19 displayed increased platelet binding and hyperresponsiveness to P-selectin and fibrinogen with respect to tumor necrosis factor-α and interleukin-1β secretion. Platelets were able to orchestrate monocyte responses driving tissue factor (TF) expression, inflammatory activation, and inflammatory cytokines secretion in SARS-CoV-2 infection. Platelet-monocyte interactions ex vivo and in SARS-CoV-2 infection model in vitro reciprocally activated monocytes and platelets, inducing the heightened secretion of a wide panel of inflammatory mediators. We identified platelet adhesion as a primary signaling mechanism inducing mediator secretion and TF expression, whereas TF signaling played major roles in amplifying inflammation by inducing proinflammatory cytokines, especially tumor necrosis factor-α and interleukin-1β. Our data identify platelet-induced TF expression and activity at the crossroad of coagulation and inflammation in severe COVID-19.
Introduction
Hypercoagulability is central in pathophysiology and also a significant determinant of mortality in patients with COVID-19.1-4 Pulmonary and extrapulmonary microvascular thrombosis is associated with multiorgan failure,5-7 occurring more frequently in COVID-19 than in influenza pneumonia.7,8 Although heparin treatment may be beneficial,9 persistent hypercoagulability and thromboinflammatory tissue damage have been reported despite prophylactic anticoagulation.5,10,11 Markers of coagulation and inflammation, including d-dimers, tumor necrosis factor-α (TNF-α) and interleukin 6 (IL-6) are early predictors of respiratory distress and mortality during COVID-19.12-16 Overwhelming inflammatory activation (“cytokine storm”) may both sustain and be amplified by hypercoagulability.17,18 Nevertheless, the mechanisms of hypercoagulability in COVID-19 patients and how it is linked to hyperinflammation are still to be determined.
Platelets are blood cells classically known by their roles in thrombosis and hemostasis.19 Beyond their hemostatic activities, platelets orchestrate inflammatory response, secreting inflammatory mediators and forming heterologous aggregates with leukocytes.19-22 Activated platelets adhere to leukocytes reprogramming their cellular functions through juxtracrine signals from P-selectin and fibrinogen-bearing integrins.23-25 Severe COVID-19 evolves with platelet hyperactivity and increased platelet-monocyte, lymphocyte, and neutrophil aggregates formation.26-29 COVID-19 postmortem pathological findings show extensive areas of microvascular tissue thrombosis containing platelet-neutrophil complexes and NETosis.5,6 Intravascular and airways NETosis is associated with case severity and mortality,6 and activated platelets in COVID-19 are a determinant to NET extrusion.5,30 We have recently shown that increased platelet activation and platelet-monocyte interaction in severe COVID-19 induce pathologic expression of tissue factor (TF),26 the main trigger of coagulation activation and thrombosis.31 Interestingly, TF-expressing monocytes represent a subset of inflammatory monocytes highly expressing proinflammatory cytokines in people living with HIV,18 but the participation of platelets in this monocyte subset reprogramming remains unknown. Our central hypothesis is that platelet-induced procoagulant and proinflammatory signaling in monocytes are linked, amplifying inflammation and hypercoagulability in COVID-19.
Here, we identified platelet and monocyte activation mechanisms involved in reciprocal loops of cellular communication that feed the thromboinflammatory process in COVID-19. We report new mechanisms of platelet-monocyte signaling involving adhesion-mediated TF expression and activity, which drives activation and proinflammatory cytokine secretion in monocytes. We stablish signaling pathways linking coagulation and inflammation in severe COVID-19 by identifying novel mechanisms of thromboinflammation associated with severity and mortality in critically ill patients.
Material and methods
Human subjects
We prospectively enrolled a cohort of 68 reverse transcription polymerase chain reaction-confirmed patients with mild (n = 22) to severe (n = 46) COVID-19 and 25 SARS-CoV-2-negative control subjects. Blood was obtained from the 46 patients with severe COVID-19 within 72 hours from intensive care unit (ICU) admission in 3 reference centers (Instituto Estadual do Cérebro Paulo Niemeyer, Hospital Copa Star, and Leblon Campaign Hospital, all in Rio de Janeiro, Brazil). Severe COVID-19 was defined as those critically ill patients presenting viral pneumonia on computed tomography scan and requiring oxygen supplementation through either a nonrebreather mask or mechanical ventilation. Twenty-two outpatients presenting mild, self-limiting COVID-19 syndrome were also included. All patients had SARS-CoV-2-confirmed diagnostic through reverse transcription polymerase chain reaction of nasal swab or tracheal aspirates. Peripheral blood samples were collected from 25 SARS-CoV-2-negative control volunteers. The characteristics of mild, severe, and control participants are presented in Table 1. Patients with mild and severe COVID-19 presented differences regarding the age and the frequency of comorbidities (Table 1), which is consistent with previous reports.32-34 Subjects of older age and chronic noncommunicable diseases were also recruited in the SARS-CoV-2-negative control group to match with patients with mild and severe COVID-19, except for hypertension and diabetes (Table 1).
All ICU-admitted patients received usual supportive care for severe COVID-19, including either noninvasive oxygen supplementation (n = 16) or mechanical ventilation (n = 30) (supplemental Table 1). Clinical information from all patients with severe COVID-19 was collected using a standardized form: ISARIC/World Health Organization Clinical Characterization Protocol for Severe Emerging Infections.35 Clinical and laboratory data were prospectively recorded and the primary outcome analyzed was 28-day mortality (n = 28 survivors and 18 nonsurvivors; supplemental Table 2). Sex, age, and the frequency of comorbidities were not different between severe patients requiring mechanical ventilation or noninvasive oxygen supplementation neither between survivors and nonsurvivors (supplemental Tables 1 and 2). All clinical investigations were conducted according to the principles of the Declaration of Helsinki. The study protocol was approved by the National Review Board (Comissão Nacional de Ética em Pesquisa – CONEP 30650420.4.1001.0008), and informed consent was obtained from all participants or patients’ representatives.
Monocyte adhesion on immobilized P-selectin or fibrinogen
Monocyte adhesion assays were performed as previously described.36 Briefly, 8-well Lab-Tek plates were incubated overnight at 4°C with phosphate-buffered saline (PBS) containing recombinant human albumin, P-selectin (10 µg/mL), or fibrinogen (100 µg/mL) and then blocked with albumin (10 mg/mL) for 4 hours at room temperature. The plates were washed twice with PBS containing 0.05% Tween-20 and 3 times with PBS. Monocytes (1 × 105) from patients with severe COVID-19 or control subjects were resuspended in 100 µL of M199 containing 10 mg/mL polymyxin B, plated on the coated surfaces and incubated overnight at 37°C in a 5% CO2 atmosphere. After 12 hours postplating, the supernatants were harvested, centrifuged to remove loose cells (500 g for 15 minutes) and stored for further quantification of inflammatory mediators. Adherent cells were fixed with 4% paraformaldehyde and the nuclei were stained with DAPI (1 µg/mL) and analyzed by fluorescence microscopy.
Platelet-monocyte ex vivo interaction
To examine whether platelets from COVID-19 patients modulate thromboinflammatory responses in monocytes from healthy volunteers, purified platelets and monocytes were incubated ex vivo at 37°C in a 5% CO2 atmosphere. Each experimental point contained 2 × 105 monocytes from a patient with COVID-19 with 2 × 107 platelets from a healthy volunteer, or 2 × 105 monocytes from a healthy volunteer with 2 × 107 platelets from a patient with COVID-19. Control monocytes plus platelets from a different healthy volunteer were used as control. In selected experiments, platelet-monocyte interactions were performed in the presence of neutralizing antibodies against P-selectin (BBA30; R&D Systems) (20 µg/mL), TF (clone 10H10 or 5G9) (50 µg/mL), the anti-integrin αIIbβ3 monoclonal antibody abciximab (50 µg/mL), or isotype-matched immunoglobulin G (IgG; 50 µg/mL). Platelet-monocyte interactions were also performed in the presence of aspirin (100 µM, A5376; Sigma), clopidogrel (300 µM, PHR1431; Sigma), or dimethyl sulfoxide (DMSO; vehicle). After 0.5, 2, or 18 hours of interaction, cells were centrifuged, the supernatants were harvested, and cells were fixed with 4% paraformaldehyde for flow cytometry analysis as described in supplemental Material. The experiment was repeated using monocytes from 2 to 3 independent healthy volunteers with similar results, and a representative data from 1 of the donors is shown. Monoclonal anti-TF antibodies were kindly provided by Wolfram Ruf (Johannes Gutenberg University Medical Center, Mainz, Germany; and Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA).
Platelet-monocyte infection in vitro
SARS-CoV-2 was originally isolated from nasopharyngeal swabs of a confirmed case from Rio de Janeiro/Brazil (GenBank accession no. MT710714). The virus was amplified for 2 to 4 days in Vero E6 cell cultures in high-glucose Dulbecco Modified Eagle’s Medium supplemented with 2% fetal bovine serum at 37°C in 5% CO2 atmosphere. Virus titers were determined by the tissue culture infectious dose at 50% and the virus stocks kept in −80°C freezers until use. All procedures involving SARS-CoV-2 culture were performed in a biosafety level 3 facility. Platelets (2 × 107) and monocytes (2 × 105) were infected with SARS-CoV-2 separately or in combination (multiplicity of infection = 0.01 virus per monocyte). In selected experiments, platelet-monocyte cocultures were infected in the presence of abciximab, anti-TF antibodies (clone 10H10 or 5G9), or isotype-matched IgG (50 µg/mL), or the PAR-1 inhibitor SCH79797 (5 µM, Tocris 1592), PAR-2 inhibitor AZ3451 (10 µM, Sigma SML2050), or DMSO (vehicle). After 12 hours of infection, supernatants were harvested and stored for future analysis, and cells were fixed with 4% paraformaldehyde for flow cytometry analysis as described in supplemental Material.
Statistical analysis
Statistics were performed using GraphPad Prism software version 7. All the numerical variables were tested regarding their distribution using the Shapiro-Wilk test. One-way analysis of variance was used to compare differences among 3 or more groups following a normal (parametric) distribution, and Tukey post hoc test was used to locate the differences between the groups. Comparisons between 2 groups were performed using the Student t test for parametric distributions or the Mann-Whitney U test for nonparametric distributions.
Results
Platelet-monocyte interaction associates with monocyte activation and immune dysfunction in COVID-19
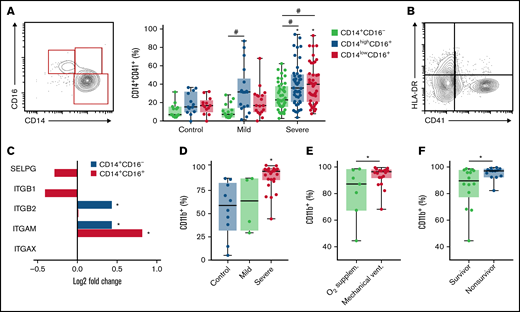
We have recently described novel mechanisms of platelet activation and platelet-induced monocyte TF expression that were associated with hypercoagulability and mortality in patients with severe COVID-19.26 We then investigated the relationship of platelet-monocyte aggregate formation and monocyte inflammatory phenotypes during severe COVID-19. Interaction with platelets was assessed by the expression of the platelet marker CD41 on the classical (CD14+CD16−), intermediate (CD14highCD16+), and nonclassical (CD14lowCD16+) monocyte subsets. As shown in Figure 1A, patients with COVID-19 presented increased levels of platelet-monocyte aggregates specifically in CD16+ intermediate and nonclassical monocytes. In addition, platelet-monocyte aggregates formed preferentially with HLA-DR-negative monocytes (Figure 2B; supplemental Figure 1). These data highlight a strong association of platelet-monocyte aggregate formation with monocyte inflammatory activation and immune dysfunction in severe COVID-19.
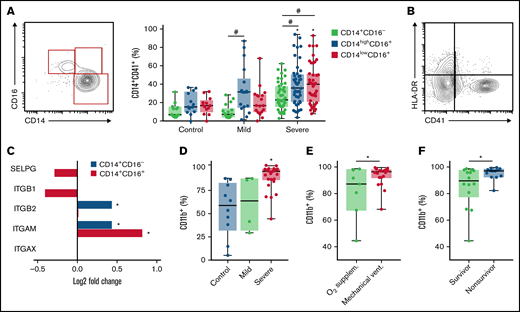
Platelet monocyte interaction associate with monocyte inflammatory activation in COVID-19. (A) The percentage of platelet-monocyte complexes among classical, intermediate, and nonclassical monocyte subsets from SARS-CoV-2-negative control participants and patients with mild to severe COVID-19 syndrome. (B) The percentage of platelet-monocyte complexes in HLA-DR-positive or negative monocytes from patients with severe COVID-19. (C) The Log2 fold change of the transcripts for P-selectin and fibrinogen receptors P-selectin glycoprotein ligand 1 (SELPG), integrin β1 (ITGB1), integrin β2 (ITGB2), integrin αX (ITGAX), and integrin αM (ITGAM) in monocytes from patients with severe COVID-19. *P < 2.5 × 10−13. (D-F) The percentage of CD11b-positive monocytes in blood from SARS-CoV-2-negative control participants and patients with mild to severe COVID-19 syndrome (D); or from patients with severe COVID-19 stratified according to the requirement of invasive mechanical ventilation or noninvasive O2 supplementation (E) or the 28-day mortality outcome as survivors or nonsurvivors (F). The horizontal lines in the box plots represent the median, the box edges represent the interquartile ranges, and the whiskers indicate the minimal and maximal value in each group. *P < .05 compared with control in the same monocyte subset; #P < .05 between selected groups.
Platelet monocyte interaction associate with monocyte inflammatory activation in COVID-19. (A) The percentage of platelet-monocyte complexes among classical, intermediate, and nonclassical monocyte subsets from SARS-CoV-2-negative control participants and patients with mild to severe COVID-19 syndrome. (B) The percentage of platelet-monocyte complexes in HLA-DR-positive or negative monocytes from patients with severe COVID-19. (C) The Log2 fold change of the transcripts for P-selectin and fibrinogen receptors P-selectin glycoprotein ligand 1 (SELPG), integrin β1 (ITGB1), integrin β2 (ITGB2), integrin αX (ITGAX), and integrin αM (ITGAM) in monocytes from patients with severe COVID-19. *P < 2.5 × 10−13. (D-F) The percentage of CD11b-positive monocytes in blood from SARS-CoV-2-negative control participants and patients with mild to severe COVID-19 syndrome (D); or from patients with severe COVID-19 stratified according to the requirement of invasive mechanical ventilation or noninvasive O2 supplementation (E) or the 28-day mortality outcome as survivors or nonsurvivors (F). The horizontal lines in the box plots represent the median, the box edges represent the interquartile ranges, and the whiskers indicate the minimal and maximal value in each group. *P < .05 compared with control in the same monocyte subset; #P < .05 between selected groups.
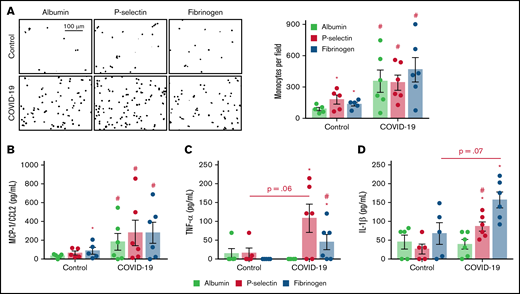
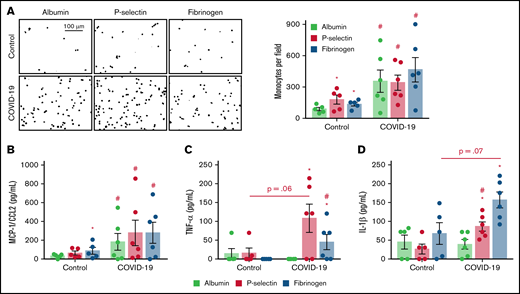
Monocytes from patients with severe COVID-19 are hyperresponsive to P-selectin and fibrinogen regarding inflammatory cytokine secretion. Monocytes (1 × 105) from patients with severe COVID-19 or control participants were plated on recombinant human albumin, P-selectin, or fibrinogen coated surfaces. (A) The number of monocytes (DAPI, nuclei) adhered on each condition is shown. Scale bar represents 100 µm. (B-D) The concentration of MCP-1/CCL2 (B), TNF-α (C), and IL-1β in each condition (D). Bars represent mean ± standard error of the mean of monocytes from 5 independent control participants and 6 independent patients with severe COVID-19. #P < .05 compared with monocytes from control participants in the same condition; *P < .05 compared with albumin.
Monocytes from patients with severe COVID-19 are hyperresponsive to P-selectin and fibrinogen regarding inflammatory cytokine secretion. Monocytes (1 × 105) from patients with severe COVID-19 or control participants were plated on recombinant human albumin, P-selectin, or fibrinogen coated surfaces. (A) The number of monocytes (DAPI, nuclei) adhered on each condition is shown. Scale bar represents 100 µm. (B-D) The concentration of MCP-1/CCL2 (B), TNF-α (C), and IL-1β in each condition (D). Bars represent mean ± standard error of the mean of monocytes from 5 independent control participants and 6 independent patients with severe COVID-19. #P < .05 compared with monocytes from control participants in the same condition; *P < .05 compared with albumin.
Monocytes from patients with COVID-19 secrete proinflammatory cytokines in response to P-selectin and fibrinogen
Considering the relationship between monocyte immunoinflammatory phenotype and interaction with platelets in severe COVID-19 (Figure 1A-B), we investigated the expression of monocyte adhesion molecules that mediate platelet-leukocyte aggregate formation. Single-cell RNA analysis has shown that the fibrinogen receptor Mac-1 subunits integrin αM and integrin β2 transcripts are increased in monocytes from patients with severe COVID-19 (Figure 1C; supplemental Figure 2). We confirmed through flow cytometry that integrin αM (CD11b) expression is increased on monocytes from patients with severe COVID-19 compared with patients with mild COVID-19 or control subjects (Figure 1D), indicating increased Mac-1 expression. Importantly, Mac-1 expression was higher in mechanically ventilated patients compared with patients under noninvasive oxygen supplementation (Figure 1E) and in patients that evolved with mortality compared with hospital discharge (Figure 1F).
To gain insights on how monocytes from severe COVID-19 patients respond to the molecules that mediate platelet-monocyte aggregate formation, we performed monocyte adhesion assays on P-selectin- or fibrinogen-coated surfaces. As expected, monocytes from healthy volunteers showed increased adhesion to recombinant P-selectin and fibrinogen when compared with recombinant human albumin (Figure 2A). Monocytes from patients with severe COVID-19, on the other hand, were more adhesive and secreted higher levels of IL-6, IL-10, and MCP-1/CCL2 regardless of the surface on which they were adhered (Figure 2A-B; supplemental Figure 3A-B). Importantly, monocytes from severe COVID-19 patients were more responsive to P-selectin- and fibrinogen-coated surfaces regarding the secretion of TNF-α, IL-1β, IL-8, MIP-1α, and MIP-1β compared with control monocytes (Figure 2C-D; supplemental Figure 3D-F). These data indicate that monocytes from severe COVID-19 patients present higher responsiveness to P-selectin and fibrinogen regarding inflammatory cytokine secretion, especially TNF-α, IL-1β, and IL-8.
Platelet adhesion and induction of TF expression precede monocyte inflammatory activation
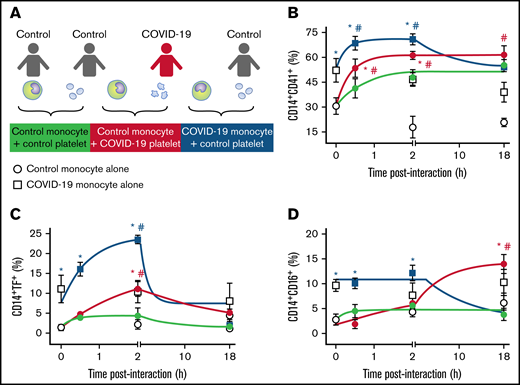
We have recently shown that activated platelets from patients with severe COVID-19 induce monocyte TF expression.26 We hypothesized that besides procoagulant pathways, platelet-monocyte interaction also orchestrates inflammation in COVID-19. To confirm this hypothesis, we incubated monocytes from healthy volunteers with platelets from patients with severe COVID-19 ex vivo (Figure 3A) and evaluated the kinetics of monocyte TF and CD16 expression. Platelets from patients with severe COVID-19 rapidly formed aggregates with control monocytes and induced TF expression up to 2 hours after interaction compared with platelets from heterologous healthy volunteers (green and red lines in Figure 3B-C). The interaction with platelets from patients with severe COVID-19 also increased CD16 expression on control monocytes, even though at a later time point (Figure 3D).
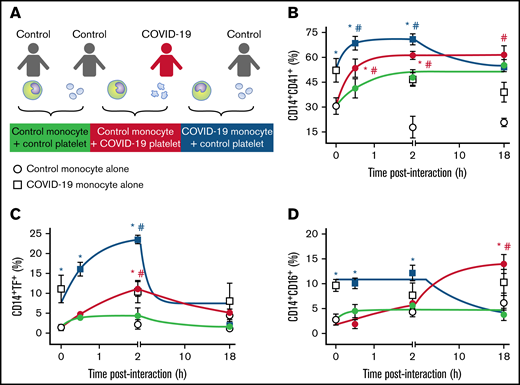
Platelet-monocyte aggregates formation, TF expression, and CD16 expression follows differential kinetics in COVID-19. (A) Monocytes from healthy volunteers (control monocyte) were incubated in the absence of platelets (open circles) or with platelets from severe COVID-19 patients (COVID-19 platelets, red circles) or from a different healthy volunteer (control platelets, gray circles) for the indicated time points. Monocytes from COVID-19 patients (COVID-19 monocyte) were also incubated in the absence of platelets (open squares) or with platelets from healthy volunteers (control platelets, black squares). The percentage of platelet-monocyte aggregates formation (B), TF-expressing monocytes (C), and CD16-positive monocytes (D) are shown. Dots represent mean ± standard error of 4 to 6 platelet and monocyte combinations from patients with COVID-19 or control participants. All experiments were repeated with cells from at least 2 independent control participants exposed to platelets or monocytes from the same patients with COVID-19 with similar results, and a representative data from 1 of the donors is shown. #P < .05 compared with baseline; *P < .05 compared with control monocytes exposed to control platelets.
Platelet-monocyte aggregates formation, TF expression, and CD16 expression follows differential kinetics in COVID-19. (A) Monocytes from healthy volunteers (control monocyte) were incubated in the absence of platelets (open circles) or with platelets from severe COVID-19 patients (COVID-19 platelets, red circles) or from a different healthy volunteer (control platelets, gray circles) for the indicated time points. Monocytes from COVID-19 patients (COVID-19 monocyte) were also incubated in the absence of platelets (open squares) or with platelets from healthy volunteers (control platelets, black squares). The percentage of platelet-monocyte aggregates formation (B), TF-expressing monocytes (C), and CD16-positive monocytes (D) are shown. Dots represent mean ± standard error of 4 to 6 platelet and monocyte combinations from patients with COVID-19 or control participants. All experiments were repeated with cells from at least 2 independent control participants exposed to platelets or monocytes from the same patients with COVID-19 with similar results, and a representative data from 1 of the donors is shown. #P < .05 compared with baseline; *P < .05 compared with control monocytes exposed to control platelets.
As previously reported, monocytes from severe COVID-19 patients present increased aggregation with platelets and higher TF expression at baseline (white symbols in Figure 3B-C).26 Interestingly, when monocytes from patients with severe COVID-19 were exposed to platelets from healthy volunteers, platelet-monocyte aggregates formation, and TF expression were further enhanced (blue lines in Figure 3B-C), indicating that platelet-monocyte aggregates from patients with severe COVID-19 recruit resting platelets to amplify TF expression. Even though the addition of control platelets potentiated aggregate formation and TF expression by COVID-19 monocytes, these were transient responses, while the interaction of control monocytes with COVID-19 platelets was sustained (Figure 3B-C). Collectively, these data suggest that platelet-mediated monocyte procoagulant and proinflammatory activation follow different kinetics and involve a complex set of signals influenced by infection-driven phenotypes of both platelets and monocytes.
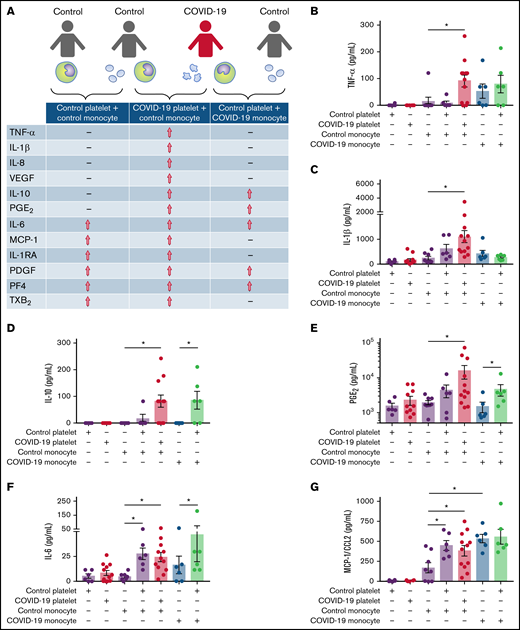
Platelet-monocyte interaction drives inflammatory mediator secretion in COVID-19
Previous studies from our group and others have demonstrated the ability of activated platelets to regulate monocyte transcription and secretion of inflammatory mediators.36,37-39 To characterize the pattern of inflammatory mediator secretion by platelet-monocyte aggregates in COVID-19, monocytes from healthy volunteers were exposed to platelets from patients with severe COVID-19 or platelets from a different healthy volunteer. Monocytes from patients with severe COVID-19 were also incubated with platelets from control participants (Figure 4A). The levels of cytokines and eicosanoids were quantified at 18 hours after interaction. As shown in Figure 4A-C and supplemental Figure 4A, increased secretion of the proinflammatory cytokines TNF-α, IL-1β, and IL-8/CXCL8 was observed in monocytes from healthy volunteers that interacted with platelets from patients with severe COVID-19, but not with control platelets. Furthermore, monocytes from healthy volunteers exposed to platelets from patients with COVID-19, or monocytes from patients with COVID-19 exposed to platelets from healthy volunteers secreted heightened levels of IL-10 and PGE2, which was not observed when control monocytes were exposed to control platelets (Figure 4D-E). Platelet-monocyte interactions also increased the secretion of the cytokines IL-1RA and IL-6, the chemokine CCL2/MCP-1, and the platelet-derived factors PF4/CXCL4 and PDGF regardless the source of the cells (from COVID-19 or from healthy donors) (Figure 4F-I; supplemental Figure 8C-D). These data highlight an inflammatory cytokine pattern that is characteristic of platelet-monocyte interactions involving platelets or monocytes from patients with COVID-19 (Figure 4A).

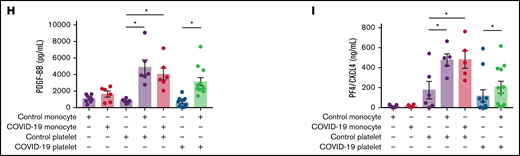
Platelet-monocyte interactions increase the secretion of inflammatory mediators in COVID-19. (A) Monocytes from healthy volunteers (control monocyte) were incubated with platelets from patients with severe COVID-19 (COVID-19 platelets) or from a different healthy volunteer (control platelets) for 18 hours, and the indicated inflammatory mediators were quantified in the supernatants. Monocytes from patients with COVID-19 (COVID-19 monocyte) were also incubated with platelets from healthy volunteers (control platelets). The concentration of TNF-α (B), IL-1β (C), IL-10 (D), PGE2 (E), IL-6 (F), MCP-1/CCL2 (G), PDGF (H), and PF4/CXCL4 (I) are shown. Bars represent mean ± standard error of the mean of 6 to 12 platelet and monocyte combinations from patients with COVID-19 or control participants. All experiments were repeated with cells from 2 independent control participants exposed to platelets or monocytes from the same patients with COVID-19 with similar results, and a representative data from 1 of the donors is shown. *P < .05 between selected groups.
Platelet-monocyte interactions increase the secretion of inflammatory mediators in COVID-19. (A) Monocytes from healthy volunteers (control monocyte) were incubated with platelets from patients with severe COVID-19 (COVID-19 platelets) or from a different healthy volunteer (control platelets) for 18 hours, and the indicated inflammatory mediators were quantified in the supernatants. Monocytes from patients with COVID-19 (COVID-19 monocyte) were also incubated with platelets from healthy volunteers (control platelets). The concentration of TNF-α (B), IL-1β (C), IL-10 (D), PGE2 (E), IL-6 (F), MCP-1/CCL2 (G), PDGF (H), and PF4/CXCL4 (I) are shown. Bars represent mean ± standard error of the mean of 6 to 12 platelet and monocyte combinations from patients with COVID-19 or control participants. All experiments were repeated with cells from 2 independent control participants exposed to platelets or monocytes from the same patients with COVID-19 with similar results, and a representative data from 1 of the donors is shown. *P < .05 between selected groups.
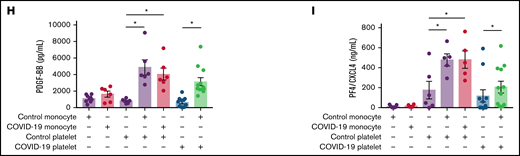
Platelets respond to SARS-CoV-2 and orchestrate monocyte activation in vitro
We next investigated the platelet and monocyte responses to SARS-CoV-2 separately and in combination. Platelets, monocytes, or platelet-monocyte cocultures (100 platelets per monocyte) were incubated with SARS-CoV-2 in vitro (multiplicity of infection, 0.01 virus per monocyte and 0.0001 virus per platelet) (Figure 5A). Platelet exposure to SARS-CoV-2 significantly increased platelet activation and secretion of granule-stored factors (Figure 5B-C). Importantly, the conjunct cytokines secreted by monocytes incubated in the presence of platelets showed increased diversity compared with monocytes infected alone (Figure 5D). Although monocytes exposed to SARS-CoV-2 alone enhanced the secretion of TNF-α, MIP-1α, and MIP-1β, monocytes incubated in the presence of platelets showed higher secretion of the inflammatory cytokines IL-1β, IL-18, and IL-1RA, and the chemokines IL-8/CXCL8, MIG/CXCL9, IP10/CXCL10, MCP-1/CCL2, and MCP-3/CCL7 (Figure 5D; supplemental Figure 9C-I). Monocytes infected in the presence of platelets also displayed increased CD16 and TF expression as compared with monocytes alone (Figure 5D). HLA-DR downregulation was a monocyte response to SARS-CoV-2 independent on the presence of platelets (Figure 5D). These data highlight platelet recognition and response to SARS-CoV-2 and platelet ability to reprogram monocyte responses to virus.

Platelets respond to SARS-CoV-2 and modulate monocytes activation in vitro. (A) Platelets, monocytes, and platelet-monocyte cocultures were kept uninfected or exposed to SARS-CoV-2 overnight. (B) The percentage of P-selectin in uninfected and SARS-CoV-2-infected platelets. (C) The fold change in platelet activation markers and mediator secretion after SARS-CoV-2 infection compared with uninfected platelets. (D) The fold change in platelet and monocyte activation markers and mediator secretion after SARS-CoV-2 infection as compared between infected and uninfected monocytes (left), infected and uninfected platelet-monocyte cocultures (middle), or in infected cocultures compared with monocytes infected alone (right). (E) The fold change in platelet activation markers and mediator secretion in SARS-CoV-2 infected cocultures compared with platelets infected alone. (F) Soluble P-selectin (sCD62P) concentration in platelets, monocytes, or platelet-monocyte cocultures after SARS-CoV-2 infection. Bars represent mean ± standard error of the mean of platelets and/or monocytes from 4 independent donors. *P < .05 compared with uninfected platelets or between selected groups.
Platelets respond to SARS-CoV-2 and modulate monocytes activation in vitro. (A) Platelets, monocytes, and platelet-monocyte cocultures were kept uninfected or exposed to SARS-CoV-2 overnight. (B) The percentage of P-selectin in uninfected and SARS-CoV-2-infected platelets. (C) The fold change in platelet activation markers and mediator secretion after SARS-CoV-2 infection compared with uninfected platelets. (D) The fold change in platelet and monocyte activation markers and mediator secretion after SARS-CoV-2 infection as compared between infected and uninfected monocytes (left), infected and uninfected platelet-monocyte cocultures (middle), or in infected cocultures compared with monocytes infected alone (right). (E) The fold change in platelet activation markers and mediator secretion in SARS-CoV-2 infected cocultures compared with platelets infected alone. (F) Soluble P-selectin (sCD62P) concentration in platelets, monocytes, or platelet-monocyte cocultures after SARS-CoV-2 infection. Bars represent mean ± standard error of the mean of platelets and/or monocytes from 4 independent donors. *P < .05 compared with uninfected platelets or between selected groups.
Platelet-monocyte interaction reciprocally activates platelets
An important step of our investigation was to examine whether platelet-monocyte interaction could also impact on the secretion of platelet-derived mediators. Interestingly, the secretion of PDGF, PF4, and TXB2, mediators produced exclusively by platelets, was increased when platelets from healthy volunteers interacted with monocytes from patients with severe COVID-19 or from different control subjects (Figure 4H-I; supplemental Figure 4D). Platelets from patients with COVID-19 were also responsive to the interaction with monocytes from healthy volunteers by releasing PF4/CXCL4 and PDGF, as compared with platelets alone (Figure 4H-I). Similarly, platelets exposed to SARS-CoV-2 in vitro in the presence of monocytes secreted higher levels of PF4/CXCL4, sCD62P, PDGF, and RANTES/CCL5 than platelets exposed to SARS-CoV-2 only (Figure 5E-F). Comparable results were observed with platelets from healthy volunteers stimulated with thrombin in vitro (supplemental Figure 5K-M), indicating that platelet activation by interaction with monocytes is not a COVID-19 exclusive feature. These data show that platelet-monocyte adhesion induces 2-way signals that affect not only the monocytes but also the platelets, increasing the secretion of stored and newly synthesized platelet factors.
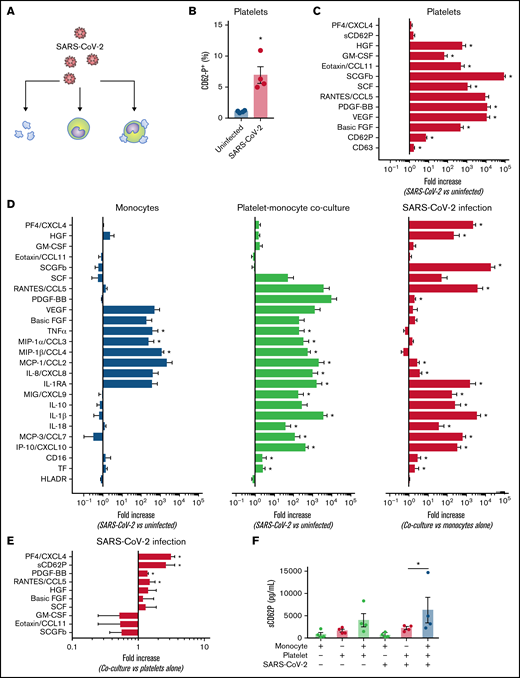
Platelets from patients with COVID-19 activate monocytes through TF-dependent and TF-independent signaling
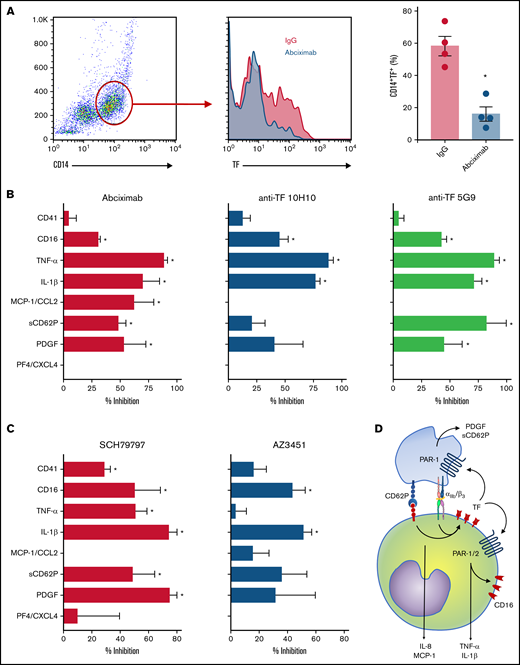
We have recently shown that P-selectin and integrin αIIb/β3 play major roles in platelet-induced TF expression in monocytes in severe COVID-19.26 Besides its roles in coagulation, monocyte TF expression and activity have been implicated in inflammatory cytokine production and immune activation.18 Considering the earlier kinetics of platelet-induced TF compared with CD16 expression on monocytes (Figure 3), we hypothesized that platelet-induced TF expression might contribute to monocyte inflammatory responses during platelet-monocyte aggregate formation. To investigate whether TF is involved on platelet-monocyte signaling, we performed ex vivo platelet-monocyte coculture in the presence of a neutralizing anti-P-selectin antibody, the anti-αIIb/β3 abciximab, and a pair of isotype-matched antibodies against distinct epitopes of TF that impair TF direct signaling (clone 10H10) or coagulation activation (clone 5G9).40 In addition, we performed ex vivo platelet-monocyte interaction in the presence of the antiplatelet drugs aspirin and clopidogrel. As shown in Figure 6A, we identified patterns of platelet-induced monocyte activation depending not only on P-selectin- and integrin αIIb/β3-mediated adhesion, but also on TF activity, leading to increased CD16 expression and TNF-α and IL-1β secretion (Figure 6A). We have also identified platelet-mediated monocyte responses depending only on P-selectin- and integrin αIIb/β3, regardless of TF activity, leading to the secretion of IL-10, IL-8/CXCL8, MIP-1α/CCL3, and MCP-1/CCL2 (Figure 6A; supplemental Figure 6). In addition, we used a pharmacological inhibitor that blocks TF coagulant and signaling activities. Exposure of control monocytes to platelets from patients with severe COVID-19 in the presence of TF inhibitor significantly impaired platelet-induced CD16 expression, and treatment with TF inhibitor completely blunted P-selectin- or fibrinogen-induced TNF-α secretion by adhered monocytes (data not shown). Even though aspirin treatment effectively inhibited platelet TXA2 synthesis and PF4 secretion, aspirin or clopidogrel was unable to impair platelet-induced monocyte activation and secretion (Figure 6B-C; supplemental Figure 7). Importantly, the secretion of the platelet-derived mediators PDGF, basic FGF, and HGF were inhibited by anti-P-selectin and/or abciximab (Figure 6A), reassuring the notion that platelet-monocyte adhesion reciprocally signals to platelets, activating platelet secretion.
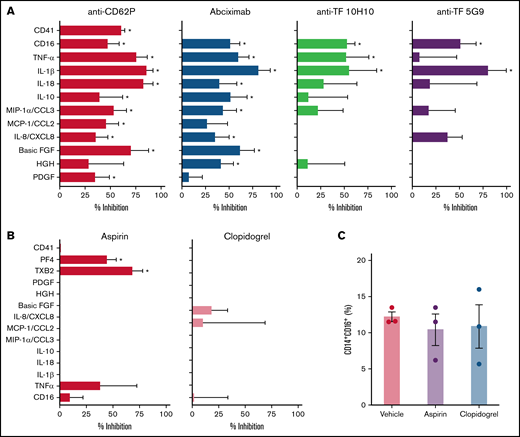
Platelets from patients with COVID-19 activate monocytes through surface interaction and TF-mediated signaling. (A) Monocytes from healthy volunteers were incubated with platelets from patients with severe COVID-19 for 18 hours in the presence of anti-P-selectin (anti-CD62P) neutralizing antibody, the anti-αIIb/β3 antibody abciximab, anti-TF clone 10H10, anti-TF clone 5G9, or isotype matched IgG. The percent inhibition on platelet-monocyte aggregate formation (CD41+ monocytes), monocyte CD16 expression, and on cytokine release is shown for each condition. (B-C) Control monocytes were exposed to platelets from patients with severe COVID-19 in the presence of the antiplatelet drugs aspirin, clopidogrel, or DMSO (vehicle). The percent inhibition on platelet-monocyte aggregate formation, monocyte CD16 expression and on cytokine release (B) and the percentage of monocytes expressing CD16 (C) are shown for each condition. Bars represent mean ± standard error of the mean of monocytes exposed to platelets from 3 to 6 independent patients with COVID-19. *P < .05 compared with isotype-matched IgG (A) or vehicle (B-C).
Platelets from patients with COVID-19 activate monocytes through surface interaction and TF-mediated signaling. (A) Monocytes from healthy volunteers were incubated with platelets from patients with severe COVID-19 for 18 hours in the presence of anti-P-selectin (anti-CD62P) neutralizing antibody, the anti-αIIb/β3 antibody abciximab, anti-TF clone 10H10, anti-TF clone 5G9, or isotype matched IgG. The percent inhibition on platelet-monocyte aggregate formation (CD41+ monocytes), monocyte CD16 expression, and on cytokine release is shown for each condition. (B-C) Control monocytes were exposed to platelets from patients with severe COVID-19 in the presence of the antiplatelet drugs aspirin, clopidogrel, or DMSO (vehicle). The percent inhibition on platelet-monocyte aggregate formation, monocyte CD16 expression and on cytokine release (B) and the percentage of monocytes expressing CD16 (C) are shown for each condition. Bars represent mean ± standard error of the mean of monocytes exposed to platelets from 3 to 6 independent patients with COVID-19. *P < .05 compared with isotype-matched IgG (A) or vehicle (B-C).
Platelet-monocyte interaction activates monocytes and platelets through TF-PAR1 and TF-PAR2 signaling
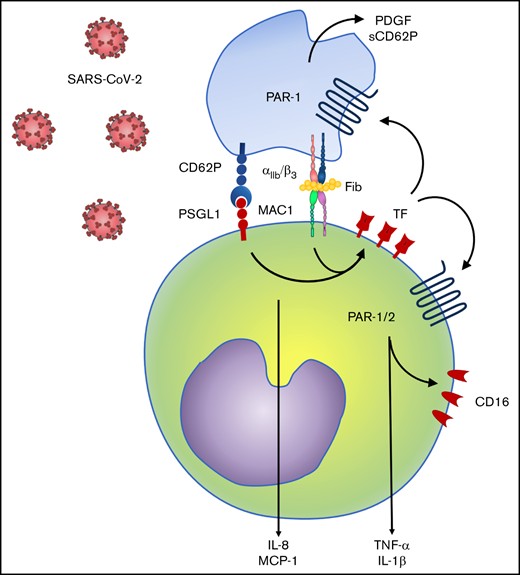
Finally, we investigated TF-mediated platelet-monocyte signaling in response to SARS-CoV-2 infection in vitro. Similar to monocytes exposed to platelets from COVID-19 patients, TF expression in response to SARS-CoV-2 was dependent on integrin-mediated platelet adhesion (Figure 7A). Enhanced expression of CD16, TNF-α, and IL-1β in platelet-monocyte cocultures were dependent on both integrin αIIb/β3 and TF-dependent signaling, whereas MCP-1/CCL2 secretion depended only on integrin signaling but not TF activity (Figure 7B). The secretion of the platelet-derived mediators PDGF and sCD62P was also inhibited by blocking the integrin αIIb/β3 and TF coagulation activity with anti-TF 5G9 clone (Figure 7B). To gain insights on the mechanisms of TF-mediated signaling in platelet-monocyte interaction, we exposed platelet-monocyte cocultures to SARS-CoV-2 in the presence of the PAR1 and PAR2 selective inhibitors SCH79797 and AZ3451, respectively. As shown in Figure 7C, monocyte activation and proinflammatory cytokine secretion depended majorly on PAR1, whereas CD16 expression and IL-1β secretion also dependent on PAR2 activation. Importantly, PAR-1 inhibition also reduced platelet-monocyte aggregate formation (CD41+ monocytes), PDGF and sCD62P secretion, indicating a role in platelet activation (Figure 7C). Collectively, these data dissect novel pathways of platelet-delivered proinflammatory signaling to monocytes through P-selectin- and integrin αIIb/β3, that amplifies platelet and monocyte activation by driving TF expression and signaling through PAR1 and 2 (Figure 7D).
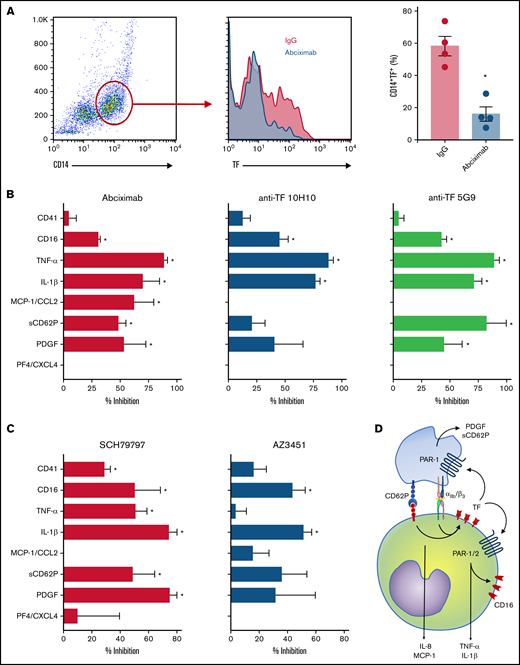
Platelet-monocyte interaction induces monocyte and platelet activation through TF-dependent PAR signaling. Platelet-monocyte cocultures were exposed to SARS-CoV-2 overnight in the presence of the anti-αIIb/β3 antibody abciximab, anti-TF clone 10H10, anti-TF clone 5G9, or isotype matched IgG. (A) The percentage of monocytes expressing TF in platelet-monocyte cocultures exposed SARS-CoV-2 in the presence of abciximab or isotype control IgG. (B) The percent inhibition on platelet-monocyte aggregate formation (CD41+ monocytes), monocyte CD16 expression, and cytokine release from platelets and monocytes is shown for each condition. (C) Platelet-monocyte cocultures were exposed to SARS-CoV-2 overnight in the presence the PAR1 inhibitor SCH79797, the PAR2 inhibitor AZ3451, or DMSO (vehicle). The percent inhibition on platelet-monocyte aggregate formation, monocyte CD16 expression, and cytokine release from platelets and monocytes is shown for each condition. (D) Schematic representation of platelet-monocyte signaling through P-selectin and integrin αIIb/β3 surface interaction and TF-mediated inflammatory amplification through PAR1 and PAR2 during severe COVID-19.
Platelet-monocyte interaction induces monocyte and platelet activation through TF-dependent PAR signaling. Platelet-monocyte cocultures were exposed to SARS-CoV-2 overnight in the presence of the anti-αIIb/β3 antibody abciximab, anti-TF clone 10H10, anti-TF clone 5G9, or isotype matched IgG. (A) The percentage of monocytes expressing TF in platelet-monocyte cocultures exposed SARS-CoV-2 in the presence of abciximab or isotype control IgG. (B) The percent inhibition on platelet-monocyte aggregate formation (CD41+ monocytes), monocyte CD16 expression, and cytokine release from platelets and monocytes is shown for each condition. (C) Platelet-monocyte cocultures were exposed to SARS-CoV-2 overnight in the presence the PAR1 inhibitor SCH79797, the PAR2 inhibitor AZ3451, or DMSO (vehicle). The percent inhibition on platelet-monocyte aggregate formation, monocyte CD16 expression, and cytokine release from platelets and monocytes is shown for each condition. (D) Schematic representation of platelet-monocyte signaling through P-selectin and integrin αIIb/β3 surface interaction and TF-mediated inflammatory amplification through PAR1 and PAR2 during severe COVID-19.
Discussion
A state of hypercoagulability with high frequency of thromboembolic complications has emerged as a key pathological feature of COVID-19.2,3 Even though coagulation disturbances are common features of critically ill patients, their frequencies are particularly higher in severe COVID-19.1,7,12,41 -44 Histopathological analysis of COVID-19 deaths or nonhuman primate infection models have revealed lung thromboinflammatory features including neutrophil and macrophage infiltration, NET-containing pulmonary microvascular thrombosis, and endothelial inflammation with platelet-fibrin deposition.5-7,45-47 These thromboinflammatory vascular occlusions are almost 10 times increased in lungs from COVID-19 fatalities compared with those from influenza pneumonia.7,8,47 Importantly, interaction with platelets is key for monocyte and neutrophil thromboinflammatory activities in COVID-19, including in driving TF expression contributing to hypercoagulability state.5,26,30 Here, we provide novel evidence of a platelet-induced proinflammatory amplification program in monocytes through adhesion molecules and TF-dependent signaling. Moreover, activated monocytes from patients with COVID-19 recruit and activate platelets, consistent with a dysregulated amplification loop that is associated with severity and mortality in COVID-19 patients.
Immune profiling of patients with severe COVID-19 has revealed an expansion of intermediate and nonclassical monocytes that fail to engage the adaptive immunity because of lower HLA-DR expression.12,48,49 This monocyte inflammatory program was also associated with poor outcomes in the patients with COVID-19 in our cohort (data not shown). Our findings support the idea that these monocyte subsets are the ones preferentially interacting with platelets during severe COVID-19. Consistently, combined single-cell transcriptome and surface proteome approaches have shown the expansion of CD16+ nonclassical monocytes highly expressing complement and type I IFN transcripts and forming aggregates with platelets.50 By single-cell RNA-sequencing and flow cytometric analysis, we identified monocytes highly expressing the leukocyte integrin Mac-1 heterodimer, a fibrinogen receptor that participates in platelet-monocyte aggregate formation.16 These monocyte phenotypic changes may explain the increased affinity to platelets and higher responsiveness to P-selectin and fibrinogen during severe COVID-19, leading to TF expression and proinflammatory cytokines secretion.
Although monocytes from patients with severe COVID-19 are highly responsive to platelets, the platelet activation status in COVID-19 also contributes to interaction with monocytes leading to TF expression and inflammatory activation. We and others have previously reported the ability of activated platelets to modulate monocyte secretion.36,37-39 We have demonstrated beforehand that platelet-monocyte aggregates formation reprogram monocyte cytokine production in dengue.36,39 We now report similar results in monocytes interacting with platelets in SARS-CoV-2 infection, except for a more proinflammatory profile marked by higher levels of TNF-α and IL-1β. Interestingly, differential signaling was required for the secretion of distinct cytokines and chemokines in this model. Platelet adhesion through P-selectin and integrin αIIb/β3 is a primary signal for the secretion of a wide range of mediators, including pro- and anti-inflammatory cytokines and chemokines and monocyte TF expression. TF, in its turn, signals to foster monocyte CD16 expression and pro-inflammatory cytokine production through PAR1 and PAR2 activation. TF-dependent PAR1 signaling was also involved in platelet activation during platelet-monocyte aggregate formation (Figure 7D). In addition to platelet-monocyte interaction, proinflammatory factors may also contribute to hyperinflammation and hypercoagulability, including in driving TF expression in monocytes.17,18 Our data describe complex mechanisms of platelet-monocyte interaction that depend on contact-mediated signaling and are amplified by TF-driven inflammatory signaling.
The mechanisms underlying platelet activation in severe COVID-19 are not yet completely understood. Our data demonstrate that platelets are responsive to SARS-CoV-2 in vitro, even though to a lower extent compared with platelets from patients with COVID-19. Of note, SARS-CoV-2 RNA, proteins and virions have been detected in platelets from infected patients, indicating the feasibility of SARS-CoV-2-induced platelet activation in natural infections.51-53 Previous studies have described that exposure to SARS-CoV-2 activates platelets in vitro,54,55 which may involve canonical interaction through angiotensin-converting enzyme 2,53,54 but also alternative receptors as CD42 and CD147.53,56,57 Similar to previously reported observations,58,59 monocytes were also responsive to SARS-CoV-2 in vitro in the present work. Importantly, our experiments revealed that monocytes infected together with platelets display amplified inflammatory activation and secrete higher levels of inflammatory cytokines. Reports from our group and others have indicated that the inflammatory mediators in the plasma of patients with COVID-19 also activate platelets.26,28,60 In whole blood from healthy volunteers reconstituted with COVID-19 plasma, IL-6 receptor blocking by tocilizumab inhibits platelet activation, platelet-leukocyte aggregates formation, and TF expression.28 Therefore, viral and inflammatory factors clearly contribute to platelet activation, which in turns amplifies inflammation in COVID-19 by reprogramming monocyte responses. We show that platelet-monocyte interaction activates both platelets and monocytes through mechanisms requiring TF-mediated PAR1 and PAR2 signaling, feeding hyperinflammation and hypercoagulability in a reciprocal amplification loop.
In summary, we describe a monocyte proinflammatory program depending on platelet-induced TF-mediated signaling during COVID-19. These platelet-monocyte responses were associated with severity and mortality in a cohort of ICU-admitted patients. However, new studies are still necessary to unravel the clinical relevance of these mechanisms. Based on the potential involvement of these cellular and molecular events in pathophysiological mechanisms of hyperinflammation and hypercoagulability, TF-mediated initiation of coagulation and inflammatory signaling may represent a target for therapeutic intervention in future clinical research in COVID-19.
Acknowledgments
The authors thank Guy A. Zimmerman for insightful discussions and comments on the manuscript, the multiuser facility on multiplex analysis and confocal imaging facility from the Rede de Plataformas Tecnológicas FIOCRUZ, and Edson F. Assis for technical assistance. The authors thank Wolfram Ruf for the anti-TF monoclonal antibodies.
This work was supported by grants from Inova Fiocruz, Fundação de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (P.T.B., F.A.B., T.M.L.S., and E.D.H.). The funders had no involvement in study design, in the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
Authorship
Contribution: E.D.H. performed experimental design and execution, data analyses, and manuscript writing; R.M.-G., L.P., I.G.A.-Q., M.M.d.C., C.Q.S., J.R.T., V.C.S., S.S.G.D., and L.T. performed part of the experiments and data analysis; H.I.N. and Í.C. performed data analysis; P.K. and C.R. performed patient inclusion, clinical management, clinical and laboratorial data compilation, and patient classification; B.B.A., H.I.N., R.Q.M., T.M.L.S., and F.A.B performed experimental design and manuscript reviewing; P.T.B. performed experimental design, manuscript reviewing, and directed the study; P.T.B. and E.D.H. conceptualized the study; and all authors reviewed and critically edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Eugenio D. Hottz, Laboratory of Immunothrombosis, Department of Biochemistry, Federal University of Juiz de Fora (UFJF), Juiz de Fora, MG 36036-330, Brazil; e-mail: eugeniohottz@gmail.com or eugenio.hottz@ufjf.br; or Patrícia T. Bozza, Laboratory of Immunopharmacology, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro 21040-900, Brazil; e-mail: pbozza@ioc.fiocruz.br or pbozza@gmail.com.