

Key Points
CPT2 K79 acetylation caused by NAD+ exhaustion and Sirt3 dysfunction resulted in LCAC accumulation and platelet damage.
Blocking acylcarnitine generation with AMPK or CPT1 inhibitors, Sirt3 agonists, and antioxidants retarded platelet storage lesion.

Abstract
The short life span of platelets is a major challenge to platelet transfusion services because of the lack of effective intervention. Here, we found that the accumulation of long-chain acylcarnitines (LCACs) is responsible for mitochondrial damage and platelet storage lesion. Further studies showed that the blockade of fatty acid oxidation and the activation of AMP-activated protein kinase (AMPK)/acetyl-CoA carboxylase/carnitine palmitoyltransferase 1 (CPT1) pathways that promote fatty acid metabolism are important reasons for the accumulation of LCACs. The excessive accumulation of LCACs can cause mitochondrial damage and a short life span of stored platelets. The mechanism study elucidated that NAD+ exhaustion and the subsequent decrease in sirtuin 3 (Sirt3) activity caused an increase in the level of CPT2 K79 acetylation, which is the primary cause of the blockade of fatty acid oxidation and the accumulation of LCACs. Blocking LCAC generation with the inhibitors of AMPK or CPT1, the agonists of Sirt3, and antioxidants tremendously retarded platelet storage lesion in vitro and prolonged the survival of stored platelets in vivo posttransfusion with single or combined use. In summary, we discovered that CPT2 acetylation attenuates fatty acid oxidation and exacerbates platelet storage lesion and may serve as a new target for improving platelet storage quality.
Introduction
Platelets are unique anucleate blood cells that are essential for normal blood clotting. The life span of human platelets is ∼7 to 10 days, and the platelet count in human peripheral blood under normal conditions is ∼100 to 300 × 109 platelets/L.1 It is very important to maintain a certain number of platelets in peripheral blood. A platelet count <20 × 109/L will cause severe life-threatening bleeding, whereas trauma, surgery, leukemia, bone marrow suppression after chemotherapy or radiotherapy, aplastic anemia, uremia, infection, immune thrombocytopenia, and other hemorrhagic diseases are frequently associated with thrombocytopenia.2 The transfusion of platelets for both prophylaxis and treatment of bleeding is relevant to all areas of medicine and surgery, and the clinical demand is huge.
Most of the platelets transfused currently are collected from healthy volunteers by using apheresis technology. A single donor platelet concentrate contains a minimum of 3 × 1011 platelets suspended in ∼200 mL of plasma, which is enough for 1 or more patients.3 The time consumption of quality examination and transportation makes the storage of platelets an inevitable process before transfusion to patients. Because platelets are very sensitive to hypothermia and hypoxia, clinical apheresis platelet concentrates are usually stored at room temperature (22 ± 2°C) with continuous gentle agitation in special bags that allow sufficient gas exchange with the external ambient air.4 Nonetheless, platelets can only be stored for up to 5 days due to platelet storage lesion. With the advent of pathogen reduction and detection technology that can greatly reduce the risk of bacterial contamination, platelet storage lesion has become a major challenge to improving the shelf life of platelet concentrates.5,6 Platelet storage lesion is associated with a decreased platelet survival and hemostatic activity in vivo after transfusion.7
Among the complex factors, metabolism is considered to be an important event in platelet storage lesion.8 Several studies showed that 85% energy in ex vivo platelets is generated from oxidation metabolism.9 Glucose metabolism via glycolysis maintains the same rate regardless of the initial concentration and availability of oxygen in platelets, whereas glucose deprivation causes a significant decrease in adenosine triphosphate (ATP) levels and an increase in apoptosis in stored platelets.10 Progressive glucose metabolite lactate production during storage results in a pH decrease and is thought to be a major cause of the death of stored platelets. Although acetate has been included in platelet additive solutions to act as a substitute fuel to reduce accumulation of lactate, acetate does not significantly ameliorate platelet storage lesion. These results suggest that metabolism is a key regulator of platelet storage lesion. How metabolism regulates stored platelet survival remains unknown, however.
In this study, we systematically analyzed the dynamic changes in human platelets during routine storage and found that the accumulation of long-chain acylcarnitines (LCACs) is responsible for platelet mitochondrial damage and short life span. CPT2 K79 acetylation caused by NAD+ exhaustion and sirtuin 3 (Sirt3) dysfunction resulted in LCAC accumulation, which was enhanced by the activation of the AMP-activated protein kinase (AMPK) signaling pathway and the subsequent inhibition of acetyl-CoA carboxylase (ACC). Moreover, Sirt3 agonists, AMPK inhibitor (AMPKi), and antioxidants significantly improved platelet storage quality when applied both individually and in combination. We therefore identified abnormal fatty acid metabolism to be a major cause for storage and immune-induced platelet damage, and targeting it may provide broad strategies for thrombocytopenia therapy.
Methods
Antibodies, reagents, cell line, and mice
Detailed descriptions of antibodies, reagents, cell line, and mice are provided in the supplemental Methods.
Platelet concentrate collection and storage
Platelet concentrates were harvested from healthy blood donors by apheresis using an apheresis separator instrument (Amicus 4R4580; Fenwal, Lake Zurich, IL). Platelets were collected into gas-permeable storage bags (C6R2316; Fenwal) with anticoagulant citrate dextrose solution formula A with a horizontal agitator at 22 ± 2°C. Samples were collected by a sterile tube at the desired time points. The study was approved by the ethics committee of Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. Written informed consent was obtained from the donors.
Mouse platelet storage
Whole blood containing 0.1 μg/mL prostaglandin E1 and 1 U/mL apyrase was centrifuged at 200g for 10 minutes to obtain platelet-rich plasma (PRP). PRP was stored in a 50-mL centrifuge tube and sealed with a sterile breathable membrane. The samples were stored at 22°C and oscillated at 100 rpm on a orbital shaker with a rotational radius of 15 mm. Next, 300 µL of PRP was taken out every day, the platelet storage lesion of stored platelets was detected by flow cytometry and aggregometer, and the acylcarnitine levels in plasma were determined by liquid chromatography/tandem mass spectrometry (supplemental Methods).
Human platelet transfusion into NCG mice
Human platelets stored on day 6 were transfused into NCG mice (GemPharmatech, Nanjing, China) by tail vein injection. Whole blood was collected from the orbital venous plexus of NCG mice at 1 minute, 0.5 hour, 2 hours, 6 hours, 1 day, or 2 days. The blood was labeled with a fluorescein isothiocyanate (FITC)-conjugated anti-human CD41 monoclonal antibody and allophycocyanin (APC) labeled anti-mouse CD41 monoclonal antibody and detected by flow cytometry. The relative platelet levels were expressed by the ratio of FITC-positive cells to APC-positive populations. The data at 1 minute after transfusion were represented as 100%.
Mouse platelet transfusion
Stored mouse platelets were labeled with Sulfo-NHS-LC-Biotin (Pierce Biotechnology, Waltham, MA) and transfused into wild-type (WT) mice by tail vein injection. Whole blood was obtained from the orbital venous plexus of NCG mice at 1 minute, 1 hour, or 2 hours. The blood was labeled with an APC-labeled streptavidin and FITC-labeled anti-mouse CD41 monoclonal antibody and detected by flow cytometry. The relative platelet levels were expressed by the percentage of APC-positive cells in FITC-positive populations. The data at 1 minute after transfusion were represented as 100%.
Statistical analysis
Statistical analyses were performed by using GraphPad Prism 8 (GraphPad Software, San Diego, CA). The t test (mean and standard error of mean) were used to determine statistical significance between 2 groups. Comparisons among multiple groups were performed by using one-way analysis of variance followed by Tukey’s multiple comparison test. N represents biological replicates. P values <.0332 were considered statistically significant.
Results
Mitochondrial damage is a major profile of stored platelets
As previously reported by others,11-17 we found that stored platelets had characteristics such as the aggregation of platelets in response to collagen declined, Annexin V exposure and caspase-3 cleavage increased, mitochondrial reactive oxygen species (mtROS) accumulated, and mitochondrial membrane potential (MMP) decreased (supplemental Figure 1A-E). Electron micrographs of the mitochondria from platelets stored 4 to 6 days showed spherical structures with fewer and disarrayed cristae, a decreased electron density of the matrix, and membrane ruptures or large vacuoles (supplemental Figure 1F). Intervention of mtROS production by N-acetyl-l-cysteine (NAC), 2-mercaptoethanesulfonic acid sodium salt, and glutathione significantly improved MMP, inhibited apoptosis, and enhanced platelet activity (supplemental Figure 1G). These results indicate that mtROS accumulation and mitochondrial dysfunction may be responsible for the apoptosis and lesion of platelets during storage.
Activation of AMPK/ACC pathway and accumulation of acylcarnitines cause mitochondrial damage in stored platelets
Several studies have shown that metabolism is essential for platelet storage.9,18 However, how metabolism regulates stored platelet survival remains an open question. We found that levels of ATP declined sharply in platelets during storage (supplemental Figure 2A). The ATP synthase inhibitor oligomycin significantly accelerated platelet lesion (supplemental Figure 2B), indicating that the process of ATP generation might play a key role in the survival of stored platelets. The metabolic fates of the 2 major ATP-generation nutrients, glucose and glutamine, were first investigated in stored platelets by isotopomer spectral analysis with the tracers [U-13C6]-glucose and [U-13C5]-glutamine at the beginning of platelet storage, respectively. As reported by others,18,19 we found that glucose was mainly converted to pyruvate and lactate through glycolysis (supplemental Figure 2C), and the levels of glucose slowly decreased in stored platelets; the ratio of glucose consumption to lactate production was roughly 1:2 (supplemental Figure 2E). The level of glutamine catabolism for the tricarboxylic acid (TCA) cycle anaplerosis was much higher (supplemental Figure 2D) and was heavily consumed, especially during the first 2 days of storage (supplemental Figure 2F). However, the addition of glucose and glutamine could not alleviate the storage lesion, and excessive glucose and glutamine caused the decrease in MMP and platelet activity, respectively (supplemental Figure 2G).
In addition to glucose and glutamine, fatty acids feed the TCA cycle. Fatty acids are converted to acyl-CoAs in the cytoplasm by acyl-CoA synthetase.20 The long chain acyl-CoAs cannot pass through the mitochondrial membrane freely. They must be converted to acylcarnitines by carnitine palmitoyltransferase 1 (CPT1) located on the mitochondrial outer membrane. After entering the mitochondria, acylcarnitines are catalyzed back to acyl-CoAs by CPT2 on the mitochondrial inner membrane. acyl-CoAs enter the mitochondrial matrix and are catalyzed to form acetyl-CoA through β-oxidation for entering the TCA cycle and eventually producing ATP.21
The metabolic fate of fatty acids was also studied in stored platelets by using bovine serum albumin–bound [U-13C16]-palmitate as the tracer for isotopomer spectral analysis. The results showed that the mole percent enrichment of the TCA cycle intermediate metabolites derived from [U-13C16]-palmitate gradually decreased during platelet storage (Figure 1A), indicating that fatty acids were effectively used by fresh platelets but fatty acid β-oxidation (FAO) was hindered in stored platelets.
![Activation of AMPK/ACC pathway and accumulation of acylcarnitines caused mitochondrial damage in stored platelets. (A) 50 µM bovine serum albumin–bound [U-13C16]-palmitate and 300 µM carnitine were added to the fresh apheresis platelets, and the 13C-labeling ratio of succinate, fumarate, or malate in stored platelets was evaluated. n = 3. (B) The levels of phosphorylation of AMPKα, AMPKβ, and ACC (p-AMPKα, p-AMPKβ and p-ACC) in stored human platelets were detected by using western blot analysis. Actin was used as a loading control. (C) Plasma acylcarnitine levels of stored human platelet concentrates were detected. n = 3. (D) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 4 days with 10 µM palmitoylcarnitine or 200 µM palmitate combined with 300 µM l-carnitine. n = 3. (E) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 6 days with 4 µM AMPKi dorsomorphin 2HCl or 0.5 mM activator AICAR. n = 3. (F) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 6 days with 40 µM malonyl-CoA or 10 µM trimetazidine. n = 3. *P < .0332, **P < .0021, ***P < .0002, ****P < .0001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/17/10.1182_bloodadvances.2021006687/4/m_advancesadv2021006687f1.png?Expires=1769255301&Signature=bF4rR7tfXcJ4qXDYMoV7zz0ywx9bV8XQfXtNQEqpPnosmuDoMfB9vAA7r~uMK3kl5BmxHHCFsECFSybcXvPy3Tja-R~6ci1AYEsbzqRr2ko~6URrgl~whA3EMy~3O0odV2Ja7EJ1zwhNF1m~fEBuV8rrdQt-0xSq11r-hsq3ewkzGVOViQZVrkGfz7uvGg~7s4NitVoP0sk-62~kWHKlBoRjRFCoOJneQwTr6FtK2G7yNm45ZPn~tJkKXVEYCj9rm6SQ7-0-WccVBZDm0Z9bnOqK7kV~Qt14S9gglwPtkTTJTn5~Mgnf7PwWXNVWB0sq7Uhufq9FR81qcz~asuV8BQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Activation of AMPK/ACC pathway and accumulation of acylcarnitines caused mitochondrial damage in stored platelets. (A) 50 µM bovine serum albumin–bound [U-13C16]-palmitate and 300 µM carnitine were added to the fresh apheresis platelets, and the 13C-labeling ratio of succinate, fumarate, or malate in stored platelets was evaluated. n = 3. (B) The levels of phosphorylation of AMPKα, AMPKβ, and ACC (p-AMPKα, p-AMPKβ and p-ACC) in stored human platelets were detected by using western blot analysis. Actin was used as a loading control. (C) Plasma acylcarnitine levels of stored human platelet concentrates were detected. n = 3. (D) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 4 days with 10 µM palmitoylcarnitine or 200 µM palmitate combined with 300 µM l-carnitine. n = 3. (E) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 6 days with 4 µM AMPKi dorsomorphin 2HCl or 0.5 mM activator AICAR. n = 3. (F) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 6 days with 40 µM malonyl-CoA or 10 µM trimetazidine. n = 3. *P < .0332, **P < .0021, ***P < .0002, ****P < .0001. ns, not significant.
Activation of AMPK/ACC pathway and accumulation of acylcarnitines caused mitochondrial damage in stored platelets. (A) 50 µM bovine serum albumin–bound [U-13C16]-palmitate and 300 µM carnitine were added to the fresh apheresis platelets, and the 13C-labeling ratio of succinate, fumarate, or malate in stored platelets was evaluated. n = 3. (B) The levels of phosphorylation of AMPKα, AMPKβ, and ACC (p-AMPKα, p-AMPKβ and p-ACC) in stored human platelets were detected by using western blot analysis. Actin was used as a loading control. (C) Plasma acylcarnitine levels of stored human platelet concentrates were detected. n = 3. (D) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 4 days with 10 µM palmitoylcarnitine or 200 µM palmitate combined with 300 µM l-carnitine. n = 3. (E) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 6 days with 4 µM AMPKi dorsomorphin 2HCl or 0.5 mM activator AICAR. n = 3. (F) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 6 days with 40 µM malonyl-CoA or 10 µM trimetazidine. n = 3. *P < .0332, **P < .0021, ***P < .0002, ****P < .0001. ns, not significant.
AMPK is a sensor of cellular energy and a metabolic master switch.22,23 AMPK can activate FAO by phosphorylating and inhibiting ACC. ACC catalyzes the carboxylation of acetyl-CoA to produce malonyl-CoA, which is an inhibitor of CPT1.24 We found that the levels of phosphorylation of AMPK at Thr172 and ACC at Ser79 were significantly elevated during storage (Figure 1B).
As mentioned earlier, [U-13C16]-palmitate–derived TCA metabolites declined instead of increased; here, the levels of acylcarnitines in stored platelets were further measured. We found that the C16:0 and C18:0 acylcarnitines accumulated during storage (Figure 1C), suggesting that cytoplasmic facilitating signals of FAO were enhanced but that the pathway of FAO was blocked. In line with a previous report that acylcarnitines were associated with poor survival of stored platelets in vivo,25 we further found that the addition of palmitoylcarnitine or palmitate + carnitine greatly accelerated storage lesion (Figure 1D). We therefore speculated that the acylcarnitine accumulation was deleterious for stored platelets. The AMPKi dorsomorphin and the CPT1 inhibitor malonyl-CoA significantly slowed down platelet apoptosis and improved the quality of stored platelets. On the contrary, the AMPK activator AICAR accelerated platelet apoptosis. The downstream β-oxidation inhibitor trimetazidine had no effects on platelet apoptosis (Figure 1E-F). These results further support our notion that the enhancement of CPT1 activity by AMPK activation and acylcarnitine accumulation are responsible for platelet storage lesion. Therefore, interfering with the accumulation of acylcarnitines may be a viable method for prolonging the life span of stored platelets.
Acetylation of proteins contributes to platelet storage lesion
Acetylation and deacetylation of mitochondrial proteins regulate a large number of metabolic pathways in mitochondria.26,27 To elucidate the cause of acylcarnitine accumulation, the changes in protein acetylation in stored platelets were evaluated (Figure 2A). The acetylation proteomics data revealed that the numbers of acetylated proteins were significantly enhanced and peaked at storage day 4. Acetylated proteins were mainly located in the cytoplasm and mitochondrion (Figure 2B). Kyoto Encyclopedia of Genes and Genomes pathway analysis showed that the acetylated proteins were involved in many signaling pathways such as platelet activation, fatty acid metabolism, and propanoate metabolism and were significantly enriched on the third or fourth day of storage (Figure 2C), consistent with the time point of the rapid decline in platelet quality.
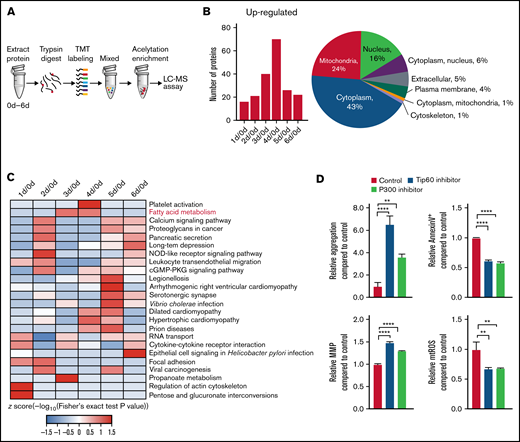
Acetylation contributes to platelet storage lesion. (A) Flow diagram for studying the dynamic changes of the acetylation level of proteins in stored platelets. The 3 × 109 platelets were collected daily from day 0 to day 6. The protein in platelets were digested by trypsin and mixed after tandem mass tag (TMT) labeling. The acetylation peptides were enriched by anti-acetylated lysine antibody conjugated beads and assayed by liquid chromatography/mass spectrometry (LC-MS). (B) The left panel shows the number of proteins in which acetylation levels were upregulated (fold ≥1.3). The right panel shows the subcellular location of the proteins in which acetylation levels were changed during storage (fold ≥1.3). (C) Kyoto Encyclopedia of Genes and Genomes pathway enrichment was performed of the proteins in which acetylation levels were changed during storage (fold ≥1.3). (D) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 6 days with 10 µM P300 inhibitor C646 or 10 µM Tip60 inhibitor MG149. **P < .0021, ****P < .0001. n = 3. cGMP-PKG, cyclic guanosine monophosphate–dependent protein kinase G.
Acetylation contributes to platelet storage lesion. (A) Flow diagram for studying the dynamic changes of the acetylation level of proteins in stored platelets. The 3 × 109 platelets were collected daily from day 0 to day 6. The protein in platelets were digested by trypsin and mixed after tandem mass tag (TMT) labeling. The acetylation peptides were enriched by anti-acetylated lysine antibody conjugated beads and assayed by liquid chromatography/mass spectrometry (LC-MS). (B) The left panel shows the number of proteins in which acetylation levels were upregulated (fold ≥1.3). The right panel shows the subcellular location of the proteins in which acetylation levels were changed during storage (fold ≥1.3). (C) Kyoto Encyclopedia of Genes and Genomes pathway enrichment was performed of the proteins in which acetylation levels were changed during storage (fold ≥1.3). (D) The aggregation, apoptosis, MMP, and mtROS levels were measured in platelets stored for 6 days with 10 µM P300 inhibitor C646 or 10 µM Tip60 inhibitor MG149. **P < .0021, ****P < .0001. n = 3. cGMP-PKG, cyclic guanosine monophosphate–dependent protein kinase G.
Acetylation was catalyzed by histone acetyltransferase. By screening the histone acetyltransferase inhibitors, we found that P300-specific inhibitor C646 and Tip60-specific inhibitor MG149 were able to improve significantly the quality of stored platelets (Figure 2D). This finding indicates that histone acetyltransferase P300 and Tip60 may catalyze the acetylation of proteins associated with platelet storage lesion.
CPT2 K79 acetylation causes acylcarnitine accumulation and mitochondrial damage
Among the acetylated proteins relevant to fatty acid metabolism, CPT2 K79 acetylation (K79ac) was confirmed by mass spectrometry (Figure 3A) and immunoblotting by a specific CPT2 K79ac antibody (Figure 3B). CPT2, as a key enzyme for β-oxidation, converts LCACs into long-chain acyl-CoAs. CPT2 mutation leads to the accumulation of LCACs, especially C16 and C18 acylcarnitine.28,29 As mentioned earlier, there was an obvious accumulation of C16:0 and C18:0 acylcarnitine during storage (Figure 1C). From these data, we inferred that CPT2 K79 acetylation is probably the major cause of acylcarnitine accumulation in stored platelets.
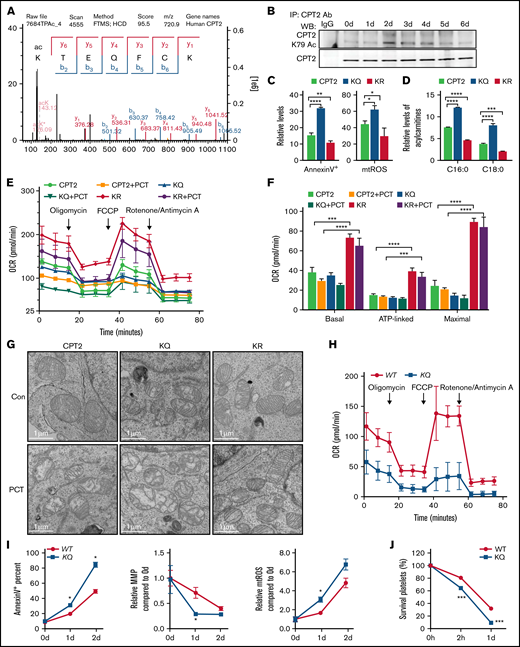
CPT2 K79 acetylation causes acylcarnitine accumulation and mitochondrial damage. (A) The mass spectrometry identification map of CPT2 K79 acetylation in stored human platelets. (B) The changes in CPT2 K79 acetylation levels in human platelets during storage were detected by western blot analysis using an anti-CPT2 K79 acetylation antibody and anti-CPT2 antibody. The specificity of this anti-CPT2 K79 acetylation antibody is evaluated in supplemental Figure 4. (C) Apoptosis and mtROS levels were detected by flow cytometry in the 293T cell line stably expressed human CPT2 (CPT2) or the 2 mutants of CPT2, K79Q (KQ) and K79R (KR). n = 3. (D) The levels of C16:0 and C18:0 acylcarnitine in CPT2, KQ, and KR 293T cells were measured. n = 3. (E) OCR was measured by Seahorse in CPT2, KQ, and KR cells or treated with 20 µM palmitoylcarnitine (PCT). (F) Basal, ATP-linked, and maximal oxygen consumption levels are represented. n = 3. (G) Representative micrograph of the mitochondria by transmission electron microscopy in CPT2, KQ, and KR cells or treated with 20 µM PCT, respectively. Scale bars represent 1 µm. (H) CPT2 K79Q mice were generated and OCR was measured in WT and KQ MEF cells using 200 µM palmitate + 300 µM l-carnitine as a substrate. (I) Apoptosis, MMP, and mtROS levels were evaluated in stored WT or KQ platelets. n = 3. (J) The survival of WT or KQ platelets stored for 1 day in vivo posttransfusion to WT mice were measured by flow cytometry. n = 3. *P < .033, **P < .0021, ***P < .0002, ****P < .0001.
CPT2 K79 acetylation causes acylcarnitine accumulation and mitochondrial damage. (A) The mass spectrometry identification map of CPT2 K79 acetylation in stored human platelets. (B) The changes in CPT2 K79 acetylation levels in human platelets during storage were detected by western blot analysis using an anti-CPT2 K79 acetylation antibody and anti-CPT2 antibody. The specificity of this anti-CPT2 K79 acetylation antibody is evaluated in supplemental Figure 4. (C) Apoptosis and mtROS levels were detected by flow cytometry in the 293T cell line stably expressed human CPT2 (CPT2) or the 2 mutants of CPT2, K79Q (KQ) and K79R (KR). n = 3. (D) The levels of C16:0 and C18:0 acylcarnitine in CPT2, KQ, and KR 293T cells were measured. n = 3. (E) OCR was measured by Seahorse in CPT2, KQ, and KR cells or treated with 20 µM palmitoylcarnitine (PCT). (F) Basal, ATP-linked, and maximal oxygen consumption levels are represented. n = 3. (G) Representative micrograph of the mitochondria by transmission electron microscopy in CPT2, KQ, and KR cells or treated with 20 µM PCT, respectively. Scale bars represent 1 µm. (H) CPT2 K79Q mice were generated and OCR was measured in WT and KQ MEF cells using 200 µM palmitate + 300 µM l-carnitine as a substrate. (I) Apoptosis, MMP, and mtROS levels were evaluated in stored WT or KQ platelets. n = 3. (J) The survival of WT or KQ platelets stored for 1 day in vivo posttransfusion to WT mice were measured by flow cytometry. n = 3. *P < .033, **P < .0021, ***P < .0002, ****P < .0001.
To further study the function of CPT2 K79 acetylation, 293T cell lines stably expressing CPT2, CPT2-K79Q as an acetyl mimic, and CPT2-K79R as an unmodified lysine mimic were constructed. The results in Figure 3C-D show that, compared with the CPT2 cells, KQ cells had much higher levels of acylcarnitines, mtROS, and apoptosis ratio, all of which were much lower in KR cells. The oxygen consumption rate (OCR) was much lower in KQ cells while much higher in KR cells compared with CPT2 cells (Figure 3E-F). Transmission electron microscopy data showed that the mitochondria in CPT2 and KR cells were healthy, with orderly arranged cristae. However, the mitochondria in KQ cells exhibited fewer and disarrayed cristae and a decreased electron density within the matrix. Moreover, KR cells were significantly resistant to the destructive effects of low concentrations of palmitoylcarnitine (Figure 3G).
To study the function of CPT2 K79 acetylation in platelet survival, CPT2 K79Q mice were further generated. The FAO was measured by evaluating the OCR of mouse embryonic fibroblasts (MEFs) with palmitate as a substrate. The OCR levels were much lower in KQ MEFs than in WT MEFs, indicating that the FAO was hindered by CPT2 KQ mutation (Figure 3H). The storage quality of platelets from WT and KQ mice was then detected. The data in Figure 3I-J show that KQ mutation significantly accelerated platelet storage lesion and promoted the clearance of stored platelets from circulation when transfused to WT mice, confirming that CPT2 K79 acetylation was detrimental to platelet storage.
These data all show that CPT2 K79 acetylation-induced acylcarnitine accumulation is a major cause of mitochondrial damage not only in platelets but also in nucleated cells.
NAD+ exhaustion–induced Sirt3 dysfunction is responsible for CPT2 K79 acetylation and acylcarnitine accumulation
Sirt3 is a major mitochondrial histone deacetylase, and its activity is dependent on NAD+ levels.30-32 We found that the levels of platelet NAD+ started to decline dramatically on the second day of storage (Figure 4A). In addition, the supplementation of NAD+ precursors nicotinamide mononucleotide (NMN) and nicotinamide riboside, and activators of nicotinamide phosphoribosyltransferase P7C3 and SBI797812, greatly reduced platelet storage lesion (Figure 4B). Correspondingly, Sirt3 activity was enhanced in stored platelets by the addition of NMN and nicotinamide riboside (supplemental Figure 3). We therefore speculated that NAD+ exhaustion may induce Sirt3 inactivation and enhancement of CPT2 K79 acetylation.
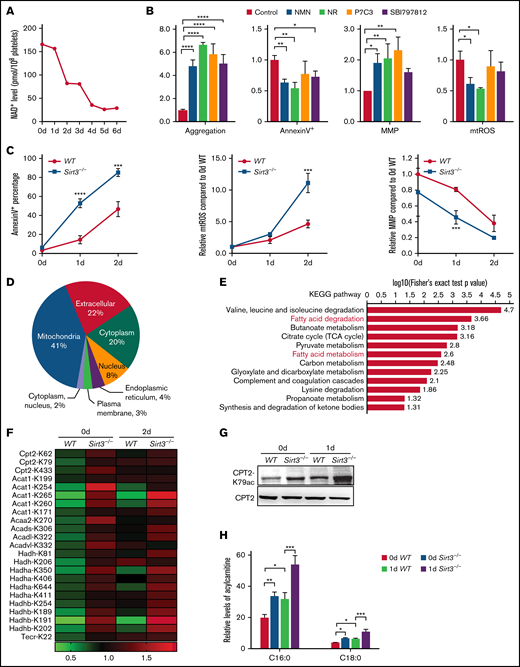
NAD+ exhaustion–induced Sirt3 dysfunction is responsible for CPT2 K79 acetylation and acylcarnitine accumulation. (A) NAD+ levels of stored human platelets were detected. (B) The aggregation, apoptosis, MMP, and mtROS levels were measured in human platelets stored for 6 days with 0.5 mM NMN, 0.5 mM nicotinamide riboside (NR), 2.5 µM P7C3, or 10 µM SBI797812. n = 3. (C) Apoptosis, mtROS levels, and MMP of stored WT and Sirt3−/− platelets were detected. n = 3. (D) Subcellular location of the proteins in which acetylation levels were changed (2-day Sirt3−/− vs 2-day WT, fold ≥1.3). (E) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of the proteins in panel D. (F) The acetylation sites of the enzymes that were involved in fatty acid metabolism are listed. (G) The levels of CPT2 K79 acetylation were detected in WT and Sirt3−/− platelets stored for 0 day and 1 day by using western blot analysis. CPT2 was used as a loading control. (H) The levels of C16:0 and C18:0 acylcarnitines were evaluated in WT and Sirt3−/− platelets stored for 0 day or 1 day, respectively. n = 3. *P < .0332, **P < .0021, ***P < .0002, ****P < .0001.
NAD+ exhaustion–induced Sirt3 dysfunction is responsible for CPT2 K79 acetylation and acylcarnitine accumulation. (A) NAD+ levels of stored human platelets were detected. (B) The aggregation, apoptosis, MMP, and mtROS levels were measured in human platelets stored for 6 days with 0.5 mM NMN, 0.5 mM nicotinamide riboside (NR), 2.5 µM P7C3, or 10 µM SBI797812. n = 3. (C) Apoptosis, mtROS levels, and MMP of stored WT and Sirt3−/− platelets were detected. n = 3. (D) Subcellular location of the proteins in which acetylation levels were changed (2-day Sirt3−/− vs 2-day WT, fold ≥1.3). (E) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of the proteins in panel D. (F) The acetylation sites of the enzymes that were involved in fatty acid metabolism are listed. (G) The levels of CPT2 K79 acetylation were detected in WT and Sirt3−/− platelets stored for 0 day and 1 day by using western blot analysis. CPT2 was used as a loading control. (H) The levels of C16:0 and C18:0 acylcarnitines were evaluated in WT and Sirt3−/− platelets stored for 0 day or 1 day, respectively. n = 3. *P < .0332, **P < .0021, ***P < .0002, ****P < .0001.
To determine the role of Sirt3 in platelet storage, the storage quality of platelets from WT and Sirt3−/− mice were compared. The data showed that Sirt3 deficiency greatly accelerated platelet storage lesion (Figure 4C). Differences in protein acetylation levels between Sirt3−/− and WT platelets were then analyzed. As expected, >40% of the proteins were located in the mitochondria (Figure 4D). Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis revealed that multiple mitochondrial metabolic pathways changed significantly in Sirt3−/− platelets, such as fatty acid degradation and metabolism (Figure 4E). Among the fatty acid metabolism pathways, the enriched key enzymes included CPT2, Acat1, Acaa2, Acads, Acadl, Acadvl, Hadh, Hadha, Hadhb, and Tecr (Figure 4F). Interestingly, the acetylation levels of CPT2 at K62, K79, and K433 were upregulated in Sirt3−/− platelets without storage, but storage only enhanced K79 acetylation significantly. The changes in CPT2 K79 acetylation in mouse platelets were also confirmed through immunoblotting (Figure 4G). Moreover, the accumulation of C16:0 and C18:0 acylcarnitines were significantly enhanced in Sirt3−/− platelets (Figure 4H). These results further support the theory that Sirt3 dysfunction is responsible for CPT2 K79 acetylation and acylcarnitine accumulation.
Combination of AMPKi, Sirt3 activator, and antioxidants significantly ameliorates the survival of stored platelets in vitro and in vivo
We found that NAD+ precursor NMN, ROS scavenger NAC, and AMPKi are capable of significantly improving the quality of stored platelets. However, the effects of the combined use of NMN, NAC, and AMPKi on platelet storage lesion is worthy of further study. The results presented in Figure 5A and supplemental Figure 5A-B show that the combination of the 3 compounds yielded much better results in improving the quality of stored platelets compared with any one of them alone.
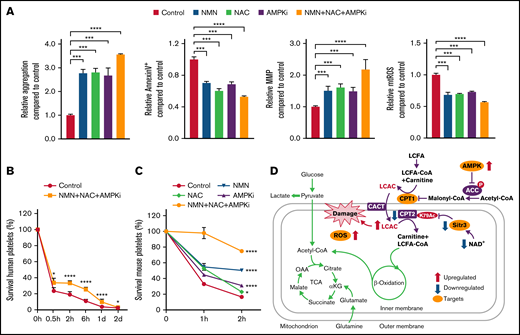
Combination of AMPKi, Sirt3 activator, and antioxidants significantly ameliorate platelet survival in vitro and in vivo. (A) The aggregation, apoptosis, MMP, and mtROS levels were measured in human platelets stored for 6 days with 0.5 mM NMN, 5 mM NAC, 4 µM AMPKi, or 0.5 mM NMN + 5 mM NAC + 4 µM AMPKi. n = 3. (B) The survival of human platelets in immunodeficient NCG mice was detected; these platelets were stored for 6 days in vitro with 0.5 mM NMN, 5 mM NAC, and 4 µM AMPKi before transfusion. n = 3. (C) The survival of mouse platelets in WT mice was detected; these platelets were stored for 2 days in vitro with 0.5 mM NMN, 5 mM NAC, 4 µM AMPKi, or the combination of 0.5 mM NMN, 5 mM NAC, and 4 µM AMPKi. n = 3. (D) Schematic model of how CPT2 K79 acetylation induces short life span of platelets. On the one hand, AMPK activation leads to increased CPT1 activity and LCAC production; on the other hand, NAD+ exhaustion and Sirt3 dysfunction results in the over-acetylation of CPT2 K79 and the decrease in CPT2 activity. The incongruous changes in CPT1 and CPT2 activities lead to the accumulation of LCAC and mitochondrial damage. Blocking LCAC generation by inhibition of AMPK or CPT1, activation of Sirt3, and antioxidants tremendously retarded platelet storage lesion by single or combined use in vitro and in vivo. *P < .0332, ***P < .0002, ****P < .0001. LCFA, long-chain fatty acid; OAA, oxaloacetate; αKG,α-ketoglutarate.
Combination of AMPKi, Sirt3 activator, and antioxidants significantly ameliorate platelet survival in vitro and in vivo. (A) The aggregation, apoptosis, MMP, and mtROS levels were measured in human platelets stored for 6 days with 0.5 mM NMN, 5 mM NAC, 4 µM AMPKi, or 0.5 mM NMN + 5 mM NAC + 4 µM AMPKi. n = 3. (B) The survival of human platelets in immunodeficient NCG mice was detected; these platelets were stored for 6 days in vitro with 0.5 mM NMN, 5 mM NAC, and 4 µM AMPKi before transfusion. n = 3. (C) The survival of mouse platelets in WT mice was detected; these platelets were stored for 2 days in vitro with 0.5 mM NMN, 5 mM NAC, 4 µM AMPKi, or the combination of 0.5 mM NMN, 5 mM NAC, and 4 µM AMPKi. n = 3. (D) Schematic model of how CPT2 K79 acetylation induces short life span of platelets. On the one hand, AMPK activation leads to increased CPT1 activity and LCAC production; on the other hand, NAD+ exhaustion and Sirt3 dysfunction results in the over-acetylation of CPT2 K79 and the decrease in CPT2 activity. The incongruous changes in CPT1 and CPT2 activities lead to the accumulation of LCAC and mitochondrial damage. Blocking LCAC generation by inhibition of AMPK or CPT1, activation of Sirt3, and antioxidants tremendously retarded platelet storage lesion by single or combined use in vitro and in vivo. *P < .0332, ***P < .0002, ****P < .0001. LCFA, long-chain fatty acid; OAA, oxaloacetate; αKG,α-ketoglutarate.
After transfusion, survival of platelets can be limited because of the rapid clearance from circulation. Upon administration of platelets, the efficacy of treatment is evaluated by posttransfusion platelet count. If the count cannot recover or drops quickly, it is likely that the transfusions may be ineffective.33,34 To evaluate the survival of stored platelets in vivo, 2 mouse transfusion models were used. In the first model, the stored human platelets were transfused to immunodeficient NCG mice.35,36 The results showed that platelet clearance was lower in the NMN + NAC + AMPKi group compared with that of the control group (Figure 5B). In the other model, the stored mouse platelets labeled by biotin were transfused to WT mice. The survival ratio of platelets stored with NMN, NAC, or AMPKi was higher than that of the control in vivo, and there was a superimposed effect for the combination (Figure 5C). These data indicate that the addition of NMN, NAC, and AMPKi could alleviate platelet storage lesion and prolong the survival period of stored platelets in vivo posttransfusion.
Cold-stored platelets are not widely used due to deep modifications in platelet shape and functionality caused by cold temperature and rapid clearance after transfusion.37,38 However, cold storage of platelets has the benefit of limiting bacterial growth that may cause infection. Furthermore, the effect of NMN, NAC, and AMPKi on cold-stored platelets was detected. We found that the platelet aggregation in response to collagen was strengthened and apoptosis was reduced in the NMN + NAC + AMPKi group (supplemental Figure 5C). The data indicate that combination of NMN, NAC, and AMPKi may also alleviate the lesion of cold-stored platelets.
Discussion
Platelet transfusions are widely used due to the great demand in clinical practices. However, platelets are not only of limited source but also face challenges in maintaining storage quality. Researchers have been focusing on the role of metabolism in platelet survival for years, but the main nutrients and metabolites closely related to platelet storage lesion remain unclear. In this study, the metabolic characteristics of glucose, glutamine, and fatty acids during platelet storage were systematically analyzed. We found that FAO kept at a low level in platelets and the ability of FAO continued to decline with the extension of storage time. It should be noted that activation of AMPK switches on FAO via phosphorylation of ACC.22-24 In contrast to the continual decline of FAO flux in mitochondria, the activation of AMPK, possibly induced by a decrease in ATP in the cytoplasm, was significantly enhanced during platelet storage. The accumulation of LCACs further verified that FAO was blocked at mitochondrial β-oxidation.
LCACs are harmful natural zwitterionic metabolites produced by the mitochondria when fatty acid metabolism is inhibited. The increase of LCAC concentration is related to the progress of various diseases, including insulin resistance, cardiovascular disease, and lung injury.39,40 In myocardial ischemia, the accumulation of LCACs is related to the increase in ROS, apoptosis, endoplasmic reticulum stress, and intracellular calcium.41 Acylcarnitines are composed of a polar head group and a hydrophobic tail. Ho et al42 showed that acylcarnitines with chain lengths longer than six carbons bind to phospholipid bilayers in the same manner as related amphipathic lipids, with the head group at the aqueous interface and the acyl chain intercalated between acyl chains of phospholipids. LCACs may play a surface active role in the cell membrane, destroying the accumulation of adjacent phospholipids, increasing the fluidity of phospholipid bilayer membrane, and leading to leakage and complete dissolution of the phospholipid bilayer.43 The longer the fatty acyl chain of LCACs, the stronger its ability to bind with phospholipids in the cell membrane, and the greater the membrane damage. LCACs can colocalize with and destroy pulmonary surfactant in vitro, reduce surface tension, and promote alveolar collapse during respiration.40 In addition, LCACs accumulate in the mitochondria of ischemic myocardium and induce the inhibition of oxidative phosphorylation, which changes the biophysical properties of the myocardial cell membrane and leads to electrophysiological disorder.43 LCACs are also thought to regulate the activities of specific ion channels and enzymes.42,44 Our data show that palmitoylcarnitine can significantly damage mitochondrial membrane morphology in both platelets and nucleated cells. We speculate that excessive accumulation of LCACs may directly damage mitochondrial structure and lead to apoptosis. However, the molecular mechanism of LCACs damage to mitochondrial structure requires further study.
LCAC accumulation indicates the possibility of insufficient CPT2 activity, which converts LCACs back to coenzyme A ester and enters β-oxidation on the luminal side of the mitochondrial inner membrane. One recent study reported that the activity and dimerization of CPT2 are directly controlled by the acetylation of K453/K457.45 However, these 2 acetylation sites were not identified in platelets. In mouse platelets, K62, K79, and K433 sites of CPT2 were acetylated, but only the K79 acetylation site was identified in human platelets. CPT2 K79Q mutation aggravated mitochondrial damage in cells and accelerated mouse platelet apoptosis, whereas K79R mutation obviously resists the mitochondrial damage induced by palmitoylcarnitine, which verified the important role of K79 acetylation in the regulation of CPT2 activity. The key sequence of mammalian CPT2 is highly conservative in mammals. The crystal structure of rat CPT2 showed that the overall structure of CPT2 was very similar to that of short-chain or medium-chain carnitine acyltransferase.46 The catalytic core of CPT2 is composed of a Y-shaped channel, which contains 3 important binding sites to recognize CoA, the palmitoyl group, and carnitine.47 In addition, CPT2 has a highly hydrophobic domain as a mitochondrial lumen membrane anchor to help the substrate palmitoylcarnitine enter the catalytic site of the CPT2 enzyme to produce palmitoyl-CoA.48 CPT2 K79 is located at a conservative QFRKTE motif, which is far from the CPT2 core catalytic region and the mitochondrial membrane anchor domain. We thus speculate that K79 acetylation affects CPT2 activity through a long-distance regulation, which requires further study.
In the past few decades, a large number of studies have attempted to find additives that can improve the quality of stored platelets. A successful example is the development of platelet additive solutions to provide extra buffering capacity. Platelet additive solution contains acetate, potassium, magnesium, and phosphate. Acetate has been proven to be a key ingredient in platelet additive solutions for providing energy directly and reducing the lactate generation.49 Here, we revealed a number of targets for interventions of platelet storage lesion, such as the elimination of mtROS to maintain mitochondria homeostasis, the inhibition of AMPK and CPT1 to block LCAC accumulation, and the activation of Sirt3 to suppress CPT2 acetylation (Figure 5D). We further showed that the combination of AMPKi, Sirt3 agonist, and mtROS scavenger is able to triple the activity of stored platelets in vitro and significantly inhibit the clearance of transfused stored platelets in vivo. In conclusion, we discovered that CPT2 acetylation and LCAC accumulation were responsible for platelet storage lesion and may serve as a new target for improving platelet storage quality.
Acknowledgments
The authors thank the support from the Core Facility of Basic Medical Sciences, Shanghai Jiao Tong University School of Medicine. They also thank Kevin Liu from SHSID for proofreading the article.
This work was supported by the National Natural Science Foundation of China (31830050, 8203000231, and 81721004, J.L.; 81970123, X.F.; 91957120, S.L.; 81800129, L.Z.; and 82070194, X.C.), National Key R&D Program of China (2021YFA0804900, J.L.) and the Innovative Research Team of High-Level Local Universities in Shanghai (SHSMU-ZDCX20211801).
Authorship
Contribution: X.F., S.L., J.L., Y.W., X.C., and J.D. designed the experiments, analyzed data, and wrote the paper; X.F., Y.W., X.C., Y.S., T.X., and L.Z. performed the experiments; and Y.X., X.W., and J.C. helped with the experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Junling Liu, Department of Biochemistry and Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Rd, Shanghai, China, 200025; e-mail: liujl@shsmu.edu.cn; Shuhai Lin, State Key Laboratory of Cellular Stress Biology, Innovation Center for Cell Signaling Network, School of Life Sciences, Xiamen University, 4221 South Xiang’an Rd, Xiamen, China, 361102; e-mail: shuhai@xmu.edu.cn; Jing Dai, Department of Laboratory Medicine, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, 197 Ruijin Second Rd, Shanghai, China, 200025; e-mail: wzdjsx@sina.com; or Lin Zhang, Department of Biochemistry and Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Rd, Shanghai, China, 200025; e-mail: linzhang3300@shsmu.edu.cn.

