

TO THE EDITOR:
GATA1 is a gene at position Xp11.23 that encodes one of the most important factors needed for hematopoiesis in both vertebrates and invertebrates.1,2 At present, several inherited mutations in exons 2 and 4 have been reported to be associated with dysregulated transcription activity, causing different pathogenic alterations and phenotypic presentations in humans.3-7 As a result of these mutations, a spectrum of diseases including hematological disorders (eg, erythroid hypoplasia with mild neutropenia and mild defects in megakaryocytopoiesis,5 severe fetal anemia with postnatal abnormalities in erythroid and platelet lineages6,8 ) and malignant hematological disorders (eg, acute megakaryocytic leukemia in children with Down syndrome9,10 ) have been described. Analysis of disease-causing GATA1 mutations clearly reflects that the location of the mutations in GATA1 determines the phenotype. In particular, for the GATA1 V205M, G208S, D218Y, and G208R mutations, studies have shown reduced affinity of GATA1 for its critical transcriptional cofactor friend of GATA1 (FOG1)6,11 -14 and reduced TAL1 complex binding of GATA1 in mutations R216Q and D218G.7,15-17 Discrepancies in the consequences on in vivo DNA binding of distinct GATA1 mutants have been noted.14 In this report, we describe a case of a patient with severe fetal anemia and the subsequent identification of a novel GATA1 mutation at p.R307H in exon 6, leading to impaired DNA binding and Ser310 phosphorylation, and offer a new link between GATA1 dysregulation and human pathology.
A 5-year-old boy with macrocytosis (mean corpuscular volume: 105 fL), elevated hemoglobin F levels (6.3%), and mild macrothrombocytopenia (thrombocytes: 121 × 103/µL) presented for further evaluation (Figure 1A). Representative bone marrow smears showed distinct dyserythropoiesis (Figure 1B-D). His medical history was remarkable for severe intrauterine anemia (minimum, 30 g/L at 21 weeks’ + 3 days’ gestation), necessitating 5 intrauterine transfusions of red cell concentrate (RCC) at 3-week intervals to achieve stable hemoglobin levels (Figure 1E). Typical morphologic anomalies associated with anemia, such as pericardial effusion, cardiomyopathy, and polyhydramnios resolved after transfusion. In standard evaluations for fetal anemia (toxicity, nutritional deficiencies, bone marrow disorders, and infectious causes) we detected no abnormalities. The birth of the patient was preterm at 36 weeks’ + 2 days’ gestation with good postnatal adaption. A single RCC was transfused at a hemoglobin level of 63 g/L, and the patient subsequently showed normal physical and mental development.
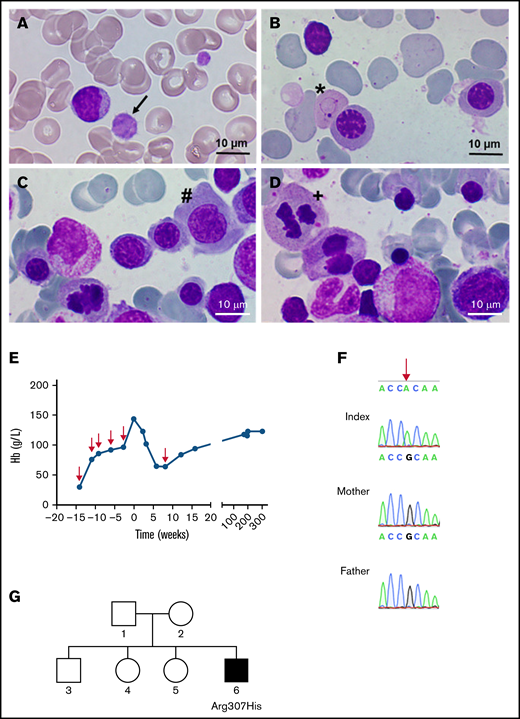
Identification of a GATA1 mutation causing severe fetal anemia. (A) The proband’s blood smear contains macrothrombocytes (black arrow). (B-D) Representative bone marrow smears show Cabot rings (B; ∗), megaloblasts (C; #), and mitosis (D; +), indicative of megaloblastic anemia. Smears were stained with May-Grünwald and photographed with a Nikon Eclipse E600 microscope equipped with a 100×/0.30 numerical aperture oil-immersion lens and a ProgRes SpeedXT core 5 camera, using Gryphax software (version 2.0). (E) Hemoglobin levels over time; red arrows indicate the time points of red blood cell transfusions. (F) DNA from the affected boy and the unaffected mother and father; the base mutation is indicated by the red arrow. (G) Pedigree of the family; the filled square represents the hemizygous male.
Identification of a GATA1 mutation causing severe fetal anemia. (A) The proband’s blood smear contains macrothrombocytes (black arrow). (B-D) Representative bone marrow smears show Cabot rings (B; ∗), megaloblasts (C; #), and mitosis (D; +), indicative of megaloblastic anemia. Smears were stained with May-Grünwald and photographed with a Nikon Eclipse E600 microscope equipped with a 100×/0.30 numerical aperture oil-immersion lens and a ProgRes SpeedXT core 5 camera, using Gryphax software (version 2.0). (E) Hemoglobin levels over time; red arrows indicate the time points of red blood cell transfusions. (F) DNA from the affected boy and the unaffected mother and father; the base mutation is indicated by the red arrow. (G) Pedigree of the family; the filled square represents the hemizygous male.
Because of the unclear origin of severe fetal anemia with sustained macrothrombocytopenia and hyperchromic macrocytosis, further genetic testing was performed. Whole-exome sequencing from an EDTA blood sample revealed hemizygosity for a novel GATA1 variant (NM_002049.4:c.920G>A, p.R307H). Among the potential findings, this variant in GATA1 was the most interesting one, based on prediction scores, allele frequency, and genotype-phenotype correlation (supplemental Table 1). The putative pathogenicity of the variant was predicted by the in silico prediction programs SIFT, MutationTaster, and PolyPhen-2, and variant frequency was obtained from the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/, accessed June 2021).18 The classification of detected variants followed the consensus recommendations of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology and revealed a combined annotation-dependent depletion score of 27.3.19 Because the mutation was classified as pathogenic, further genetic testing of the patient and both parents was performed after receipt of informed consent, in accordance with the Declaration of Helsinki and approval by the Ethics Committee of the Medical University of Vienna. Directed dye-terminator Sanger sequencing of the 5′ part (c.875-30 to c.1100) of exon 6 of GATA1 (NM_002049.4) confirmed an amino acid replacement at p.R307H (Figure 1F) in the patient only, thus confirming a de novo mutation (Figure 1G).
To evaluate the functional consequence of this mutation, transfections of erythroid cell line K562 and Jurkat TAg cells were performed. Detailed descriptions of all experiments are provided in the supplemental Methods. We first analyzed DNA binding activity, because the C-terminal zinc finger of GATA1 is known to be substantial for the predominant DNA binding activity, to enable GATA1 to associate with its (A/T)GATA(A/G) consensus sequence,8,20 such as in patients with macrothrombocytopenia, congenital porphyria, β-thalassemia, and gray platelet syndrome.7,15-17 Electrophoretic mobility shift assay (EMSA) showed that DNA binding of GATA1R307H was strongly reduced in hemin-stimulated K562 cells (Figure 2A) and comparable in Jurkat cells (supplemental Figure 1A), indicating that the R307H mutation inhibits DNA binding of GATA1 in erythroid cells. Given that other mutations (eg, V205M, G208S, and D218G)6,11-13 are known to reduce the affinity of GATA1 for FOG1 and that preclinical data have demonstrated murine Ser310 to be essential for erythroid differentiation,21 we determined the phosphorylation status on Ser310. No phosphorylation of GATA1R307H on Ser310 in K562 and Jurkat cells was observed in western blot analysis (Figure 2B; supplemental Figure 1B). According to the manufacturer, the anti–phospho-GATA1 (Ser 310) antibody was produced using as an immunogen, a peptide sequence of human GATA1 around Ser310 (K-A-S(p)-G-K) that does not include the mutation site R307H. To confirm the functional impact of reduced Ser310 phosphorylation, coimmunoprecipitation of FOG1 and GATA1 was performed. GATA1R307H showed reduced affinity to FOG1 in hemin-stimulated K562 cells (Figure 2C). The role of Ser310 phosphorylation for GATA1 affinity to FOG1 is historically controversial, depending on the experimental setup used.21-24 Nevertheless, the clinical presentation of our patient with severe fetal anemia and thrombocytopenia and impaired maturation of the erythrocytes, combined with reduced phosphorylation resulting in impaired GATA1/FOG1 binding in R307H mutation, underlines the clinical relevance. Kadri et al showed that phosphorylation at Ser310 enhances the affinity of GATA1 for its cofactor, FOG1. In turn, FOG1 displaces pRB/E2F-2 from GATA1, resulting in release of proliferative E2F-2. Mice harboring a Gata1 S310A mutation develop severe anemia when a compensatory pathway for EF-2 production (IGF1 signaling) is abolished.21 Furthermore, our patient developed severe fetal anemia and manifested with persistent anisocytosis and elevated levels of HbF during childhood (39.7% HbF at birth; 6.7% at 5 years of age). Previous studies have shown that GATA1 plays a fundamental role in fetal globin gene expression (eg, gene silencing of HBG1 and HBG2 and hemoglobin switching to HBB is promoted by GATA1).14,25-27 Finally, it is important to mention that the R307H change itself may have an effect, even if Ser310 is no longer phosphorylated. This part of GATA1 gets acetylated, and it is known to be important for in vivo DNA binding, but not in vitro.28
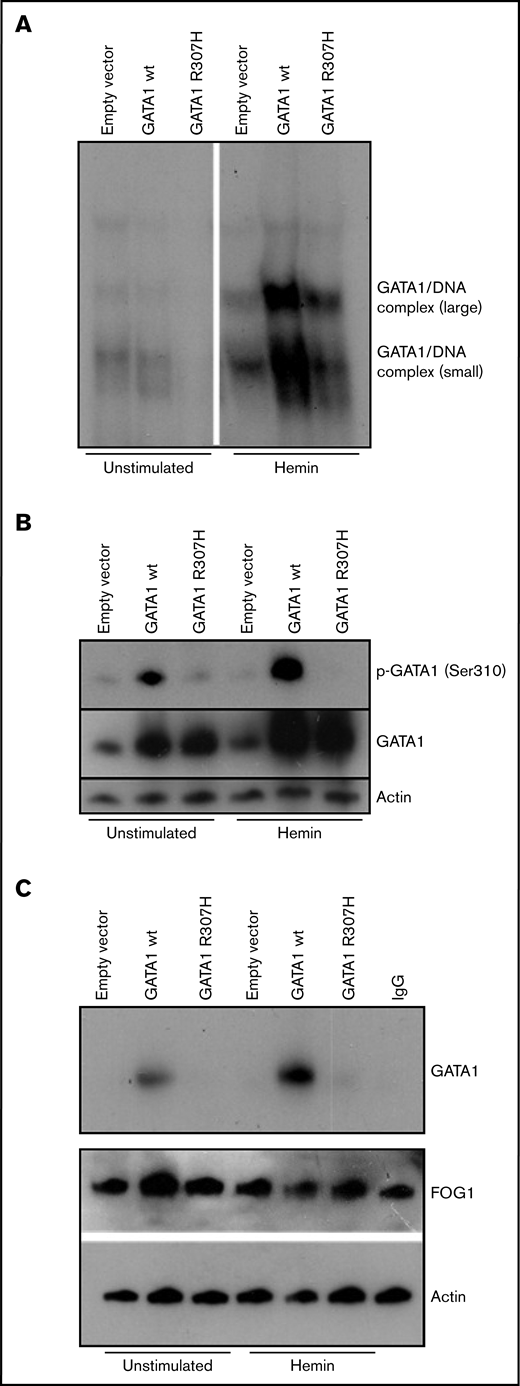
Biochemical characterization of mutant GATA1R307H. K562 cells were transfected with wild-type and mutated (c.920G>A) GATA1 cDNA constructs and stimulated for 3 days with 0.05 mM bovine hemin. (A) Nuclear extracts of wild-type and GATA1R307H-transfected cells were incubated with a 32P-labeled, double-stranded oligonucleotide probe (5′-CAC TTG ATA ACA GAA AGT GAT AAC TCT-3′). EMSA shows reduced DNA binding of GATA1R307H. (B) Immunoblot analysis of total and phosphorylated GATA1 was performed. Phosphorylation of Ser310 was totally absent in GATAR307H. (C) Immunoprecipitation of lysates with FOG1 antibody followed by western blot of GATA1. Input control of actin and FOG1 is shown. GATA1R307H showed reduced FOG1 affinity.
Biochemical characterization of mutant GATA1R307H. K562 cells were transfected with wild-type and mutated (c.920G>A) GATA1 cDNA constructs and stimulated for 3 days with 0.05 mM bovine hemin. (A) Nuclear extracts of wild-type and GATA1R307H-transfected cells were incubated with a 32P-labeled, double-stranded oligonucleotide probe (5′-CAC TTG ATA ACA GAA AGT GAT AAC TCT-3′). EMSA shows reduced DNA binding of GATA1R307H. (B) Immunoblot analysis of total and phosphorylated GATA1 was performed. Phosphorylation of Ser310 was totally absent in GATAR307H. (C) Immunoprecipitation of lysates with FOG1 antibody followed by western blot of GATA1. Input control of actin and FOG1 is shown. GATA1R307H showed reduced FOG1 affinity.
In summary, we describe a de novo mutation in GATA1R307H with the phenotypical expression of severe fetal anemia with sustained impairment of hematopoietic cell proliferation of the megakaryocytic and erythropoietic lineage. The mutated GATA1 showed impaired DNA binding, absence of Ser310 phosphorylation and reduced GATA1/FOG1 affinity, which is known to be crucial for erythroid differentiation. We conclude that recognition of this novel mutation is of critical importance in the interpretation of fetal anemia, as it can predict the impact on the course of disease and influence management in affected patients.
Acknowledgments: The authors thank Günter Weiss (Department of Internal Medicine, Medical University of Innsbruck) for the gift of K562 cells, Friedrich Fresser (Institute of Human Genetics, Medical University of Innsbruck) for cloning the GATA1R307H mutant, and Nina Posch (Institute of Cell Genetics, Medical University of Innsbruck) for transfection of the Jurkat cells.
This work was supported by grants from “Kinderkrebshilfe Tirol und Vorarlberg” and “Kinderkrebshilfe Südtirol-Regenbogen.”
Contribution: B.H., R.C., and A.M. designed the study; B.H. and R.C. collected the data; all authors analyzed the data; B.H., T.G., and R.C. wrote the manuscript; and all authors reviewed, revised, and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Roman Crazzolara, Department of Pediatrics, Medical University of Innsbruck, Anichstrasse 35, 6020 Innsbruck, Austria; e-mail: roman.crazzolara@i-med.ac.at; and Thomas Gruber, Institute of Translational Cell Genetics, Medical University of Innsbruck, Anichstrasse 35, 6020 Innsbruck, Austria; e-mail thomas.gruber@i-med.ac.at.


