TO THE EDITOR:
Pathogenic variants in each of the fibrinogen peptides resulting in quantitative or qualitative disorders have been described.1,2 Quantitative disorders include afibrinogenemia and hypofibrinogenemia. Hypofibrinogenemia may result from mutations that decrease messenger RNA production, alter messenger RNA stability, cause autophagy of misfolded proteins, or shorten fibrinogen half-life.3 Dysfibrinogenemia has been described involving mutations affecting thrombin cleavage,4 polymerization,5 lateral aggregation,6 factor XIII (FXIII) binding,7 fibrinolysis,8 rheology,9 or a combination of functional and structural abnormalities. Many fibrinogen variants are asymptomatic. Others are associated with mild to severe bleeding, thrombosis, or a combination of bleeding and thrombosis.10,11
The fibrinogen hexamer contains 29 disulfide bridges without free cysteine residues.12 Mutations affecting 1 residue of a Cys-Cys disulfide pair results in a free cysteine residue, which may form aberrant disulfide bridges with cysteines within the same fibrinogen molecule, different fibrinogen molecules, or other proteins with unpaired cysteines, such as albumin.13 Several fibrinogen variants resulting in unpaired cysteines have been described resulting in a range of phenotypes, including thrombophilia.6,8,14 -16 Here, we report the case of an adolescent female with an unprovoked portal vein thrombosis that was subsequently identified as carrying fibrinogen Villeurbanne II, a β-chain variant encoding a free cysteine (Tyr356Cys) that was previously concluded to cause hypofibrinogenemia with an absence of the variant in circulation.17 Our analyses show that fibrinogen Villeurbanne II is a hypodysfibrinogenemia that forms complexes with albumin.
The patient is a 16-year-old multiracial female with no significant past medical history who presented with 3 months of abdominal pain, nausea, and unintentional weight loss. Ultrasonography revealed a portal vein thrombosis with extension into the superior mesenteric vein. Her risk factors for thrombosis were remote and modest. Six weeks after her symptoms began, she was a restrained passenger in a motor vehicle accident. She was evaluated and treated in an emergency department and released with no evidence of significant trauma. She started combined oral contraceptives for contraception 6 months prior to presentation. There was no maternal family history of thrombosis or bleeding. Paternal family history is unknown.
At presentation, she had a prolonged prothrombin time (14 seconds; 9.6-11.6) with a normal prothrombin time mixing study, normal partial thromboplastin time (29.8 seconds; 24.3-33.6), and low fibrinogen (114 mg/dL; 200-400, Clauss method, Siemens). Thrombin and reptilase times were prolonged at diagnosis. Quantification of factors II through XI were normal except for an elevated FVIII activity (200%). Complete blood count was normal. Anticoagulation with enoxaparin was initiated. Anti-Xa levels were appropriately therapeutic. On hospital day 2, she developed severe menstrual bleeding with a 1.6 g/dL decrease in hemoglobin and a decrease in fibrinogen to <70 mg/dL. She was placed on norethindrone for menstrual suppression and switched to unfractionated heparin anticoagulation. Fibrinogen remained low throughout admission. She required weekly cryoprecipitate to maintain fibrinogen levels >80 g/dL.
She had no evidence of an inherited or acquired thrombophilia. Testing included analyses for factor V Leiden, prothrombin G20201A, antithrombin activity, protein C and protein S antigen and activity, homocysteine, lipoprotein(a), antiphospholipid antibody panel, and JAK2 mutational analyses. The patient’s urea clot solubility test was normal. Fibrinogen antigen and activity levels from several separate samples sent to Versiti Laboratory consistently revealed low fibrinogen antigen and activity levels, generally <110 g/dL. The activity:antigen ratio was sometimes <0.72, suggesting a hypodysfibrinogenemia, but was not consistently abnormal. Genetic testing via Sanger sequencing revealed a heterozygous pathogenic variant in FGB, (c.1067A>G; p.Tyr356Cys) previously reported as a hypofibrinogenemia (fibrinogen Villeurbanne II).17 No variants were noted in FGA or FGG. The patient’s mother does not carry this variant. Her father was not available for testing. The patient remains on anticoagulation and is currently on Apixaban.
The study was approved by the Institutional Review Board of Cincinnati Children’s Hospital Medical Center and informed consent was obtained.
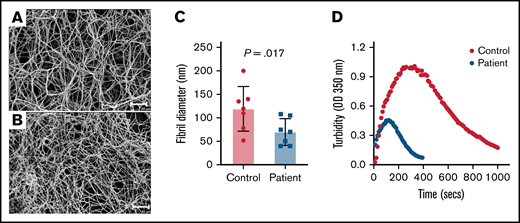
Fibrinogen was purified from plasma by ammonium sulfate precipitation as previously described.18 Fibrin clots were generated using a 1.77-mg/mL solution of purified fibrinogen by addition of 0.5 U/mL FXIIIa (HFXIIIa 1314; Enzyme Research Labs) and 0.05 U/mL thrombin (HT1002A; Enzyme Research Labs), prepared for scanning electron microscopy (SEM) as previously described.19 Three clot preparations were made from the patient’s fibrinogen, and 3 were made from control (healthy donor) fibrinogen. Two to 3 representative sections from each clot preparation were selected based on image coverage and quality and photographed using a Hitachi SU8010 electron microscope. Ten fibrils from each micrograph were randomly selected for measurement.20,21 Figure 1 represents the average diameter from each selection.

Fibrinogen Villeurbanne II forms abnormal-appearing polymer. Shown are SEM micrographs taken at the same magnification of fibrin clots produced from a 1.77-mg/mL fibrinogen solution purified from plasma obtained from a healthy donor (A) and the patient (B). Size bars, 20 μm. The patient’s fibrin clot was poorly formed, consisting of long, thin fibers with little bundling and irregularly spaced branch points. (C) Quantitative analyses of a 40-μm × 40-μm section of the micrograph show that the patient’s fibrils are significantly thinner than the control. The data represent the mean and SEM. The P value was generated using a Mann-Whitney U test. (D) Shown are the results of a turbidimetric fibrinolytic assay performed using the patient’s and control (normal) plasma. OD, optical density.
Fibrinogen Villeurbanne II forms abnormal-appearing polymer. Shown are SEM micrographs taken at the same magnification of fibrin clots produced from a 1.77-mg/mL fibrinogen solution purified from plasma obtained from a healthy donor (A) and the patient (B). Size bars, 20 μm. The patient’s fibrin clot was poorly formed, consisting of long, thin fibers with little bundling and irregularly spaced branch points. (C) Quantitative analyses of a 40-μm × 40-μm section of the micrograph show that the patient’s fibrils are significantly thinner than the control. The data represent the mean and SEM. The P value was generated using a Mann-Whitney U test. (D) Shown are the results of a turbidimetric fibrinolytic assay performed using the patient’s and control (normal) plasma. OD, optical density.
A turbidimetric clot lysis assay was performed following addition of 10 mM CaCl2 and 2 U/mL thrombin to citrated patient and control plasma in the presence of tissue plasminogen activator (9 μg/mL; Genentech). Fibrin dissolution was measured in a spectrophotometer (DU730; Life Science UV/Vis Spectrophotometer, Beckman Coulter) by change in absorbance at an optical density of 350 nm.
Plasma samples were separated on NuPAGE precast gels (NP0322BOX; Invitrogen/Life Technologies), transferred using an XCell II Blot module (#090707-098; Invitrogen/Life Technologies) onto Immobilon-FL membranes (#IPFL00010; EMD Millipore), and probed with specific primary antibodies (#sc-69775; Santa Cruz Biotechnology); albumin (#46293; Santa Cruz Biotechnology). To analyze fibrinogen interactions with albumin, we performed an immunoprecipitation for fibrinogen followed by a Western blot for albumin. Coimmunoprecipitation was performed using A/G agarose beads (sc-2003; Santa Cruz Biotechnology) and 2 μL of antibody per 1 mL of diluted plasma. The following IRDye-conjugated secondary antibodies were used: donkey anti-mouse (#926-32212; Li-Cor Biosciences) and donkey anti-goat (#926-32214; Li-Cor Biosciences).
To determine if fibrinogen Villeurbanne II alters fibrinogen polymerization, we compared SEM images of crosslinked fibrin clots formed from equal concentrations of the patient’s fibrinogen and a healthy control. The patient’s clots were poorly formed, consisting of long, thin fibers with little bundling and irregularly spaced branch points (Figure 1A). Quantitative analyses demonstrated that the patient’s fibrils were significantly thinner than control fibrils (Figure 1B), suggesting impaired lateral aggregation. To evaluate clot stability, we performed a clot lysis assay using control and patient plasma. Maximum absorbance was significantly less with patient plasma vs control plasma (Figure 1D). This was likely largely driven by the fact that fibrinogen concentration in the patient’s plasma was lower than that in control plasma. Clot lysis appeared normal in the patient’s plasma, but this needs to be interpreted with caution given differences in fibrinogen concentration. Unfortunately, we did not have sufficient sample to perform this assay using purified fibrinogen. Nevertheless, these data suggest that any potential prothrombotic effect of the mutant fibrinogen is not driven by a significant resistance to fibrinolysis.
Mutations resulting in a free-cysteine residue have been characterized in all 3 chains and are associated with dysfibrinogenemia.8,15,17,22,23 Fibrinogen variants with unpaired cysteines have been shown to form disulfide bridges with other plasma proteins, other fibrinogen molecules, or within the same fibrinogen molecule. Albumin, the most prevalent plasma protein, has an unpaired cysteine residue, making it a candidate to aggregate with the mutant fibrinogen.22,24 Western blot analyses utilizing reducing and nonreducing conditions appeared similar between the patient and control (Figure 2A). When coimmunoprecipitation followed by Western blot was performed with our patient’s fibrinogen and albumin, we observed a 66-kDa band corresponding to the molecular weight of albumin that was not observed in control plasma, suggesting an interaction between the mutant fibrinogen and albumin (Figure 2B). Notably, fibrinogen variants previously described to form disulfide bridges with albumin also displayed the formation of thin protofibrils and impaired lateral aggregation via SEM imaging, paralleling this study.16 Although we could not determine the precise concentration of mutant fibrinogen in our patient’s plasma, these data strongly suggest that Fibrinogen Villeurbanne II is present in plasma, forms disulfide bridges with albumin, and is present in sufficient quantities to impair clot formation. Recently developed time-of-flight mass spectrometry could represent an approach to accurately measure Fibrinogen Villeurbanne II in plasma.15 These findings contradict a previous description of Fibrinogen Villeurbanne II, concluding it is not present in plasma and represents a hypofibrinogenemia.17
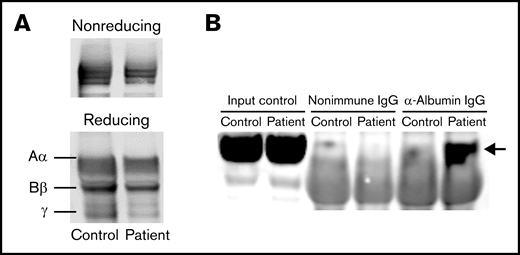
Fibrinogen Villeurbanne II forms aggregates with albumin in plasma. (A) The patient’s fibrinogen and fibrinogen from a healthy control appeared similar when blotted under reducing and nonreducing conditions. (B) Western blot analyses of fibrinogen coimmunoprecipitation demonstrating an interaction between the patient’s fibrinogen and albumin (at 66 kDa; arrow) that was not observed in fibrinogen from a healthy control, suggesting that the mutant fibrinogen forms aggregates with albumin. IgG, immunoglobulin G.
Fibrinogen Villeurbanne II forms aggregates with albumin in plasma. (A) The patient’s fibrinogen and fibrinogen from a healthy control appeared similar when blotted under reducing and nonreducing conditions. (B) Western blot analyses of fibrinogen coimmunoprecipitation demonstrating an interaction between the patient’s fibrinogen and albumin (at 66 kDa; arrow) that was not observed in fibrinogen from a healthy control, suggesting that the mutant fibrinogen forms aggregates with albumin. IgG, immunoglobulin G.
The clinical distinction between hypofibrinogenemia and hypodysfibrinogenemia is important, as the evidence suggests hypodysfibrinogenemia may confer bleeding and thrombotic risks.11 Genotype-phenotype correlations are difficult to establish because of the high allelic heterogeneity of hypodysfibrinogenemia and its relative rarity.25 In 1 review of 51 cases of hypodysfibrinogenemia consisting of 32 causative mutations, 22% were asymptomatic at diagnosis; 45% reported abnormal bleeding; 43% had a history of thrombosis; and 9.8% had both abnormal bleeding and thrombosis.10 Given our patient’s presentation with an unprovoked portal vein thrombosis, Fibrinogen Villeurbanne II may predispose to thrombosis in addition to bleeding.
Contribution: B.J.F. designed research, performed research, analyzed data, and wrote the paper; B.K.S. designed research and performed research; H.M.R. designed research and performed research; L.R. performed research; A.P.O. analyzed data and provided expert insight; M.J.F. analyzed data, provided expert insight, and edited the manuscript; E.S.M. analyzed data, provided expert insight, and edited the manuscript; and J.P. designed research, analyzed data, provided expert insight, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Brenton J. Francisco, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1208, New York, NY 10029-6574; e-mail: brenton.francisco@mssm.edu.