

Key Points
With >3 years median follow-up, D-RVd achieved durable responses that further deepened with D-R maintenance in the safety run-in cohort.
No new safety concerns were identified with D-RVd after longer follow-up in the safety run-in cohort.
Abstract
The phase 2 GRIFFIN study of daratumumab plus lenalidomide/bortezomib/dexamethasone (D-RVd) for transplant-eligible, newly diagnosed multiple myeloma included a safety run-in phase followed by a randomized phase. The ongoing randomized phase has met its prespecified primary end point of an improved stringent complete response (sCR) rate after consolidation for D-RVd (reported elsewhere). Final analysis of the safety run-in cohort is reported herein and provides longer follow-up (median, 40.8 months) encompassing daratumumab plus lenalidomide (D-R) maintenance therapy. Patients in the safety run-in cohort (N = 16) received 4 induction cycles (D-RVd), high-dose melphalan supported by autologous stem cell transplant, 2 consolidation cycles (D-RVd), and 24 months of maintenance (D-R). By the end of consolidation, all patients had responded, with a best response of sCR in 9 (56.3%) patients; 8 (50.0%) patients were minimal residual disease (MRD) negative (10‒5 threshold). After maintenance, 15 (93.8%) patients had achieved a best response of sCR, and 13 (81.3%) patients were MRD (10‒5) negative. Estimated 36-month progression-free and overall survival rates were 78.1% and 93.8%, respectively. One death from progressive disease occurred in the patient who did not achieve sCR. Observed safety profiles were consistent with daratumumab and RVd. With >3 years of median follow-up, D-RVd achieved durable responses that deepened with D-R maintenance. This study was registered at www.clinicaltrials.gov as #NCT02874742.
Introduction
Daratumumab, a human IgGκ monoclonal antibody, targets CD38 with a direct on-tumor1-4 and immunomodulatory5-7 mechanism of action. Based on the phase 3 CASSIOPEIA study (www.clinicaltrials.gov, #NCT02541383),8 daratumumab plus bortezomib/thalidomide/dexamethasone (VTd) was approved for treatment of transplant-eligible, newly diagnosed multiple myeloma (NDMM).9,10
In the United States, the standard of care for transplant-eligible NDMM is lenalidomide/bortezomib/dexamethasone (RVd), rather than VTd.11 Thus, the phase 2 GRIFFIN study was designed to assess daratumumab plus RVd (D-RVd) for transplant-eligible NDMM and included a safety run-in phase followed by a randomized phase.12,13 After the safety run-in phase (N = 16), the independent data review committee recommended proceeding to the randomized phase, as no patients discontinued treatment because of dose-limiting toxicities (DLTs).12 The randomized phase (N = 207) met the prespecified primary end point, with an improved rate of stringent complete response (sCR) by the end of consolidation (D-RVd, 42.4% vs RVd, 32.0%; 1-sided P = .068; median follow-up, 13.5 months). With additional daratumumab plus lenalidomide (D-R) or lenalidomide maintenance therapy, responses continued to deepen and remained higher for D-RVd vs RVd as of the last follow-up.13
We report the final analysis of the GRIFFIN safety run-in cohort after completion of D-RVd treatment and 24 months of D-R maintenance therapy.
Methods
Patients with NDMM eligible for high-dose therapy (HDT) and autologous stem cell transplant (ASCT) were enrolled. Complete eligibility criteria have been published.13 Patients in the safety run-in cohort received four 21-day induction cycles (D-RVd), high-dose therapy (melphalan), ASCT, two 21-day consolidation cycles (D-RVd), and twenty-six 28-day maintenance cycles (D-R). During induction and consolidation, the patients received lenalidomide 25 mg orally on days 1 to 14; bortezomib 1.3 mg/m2 subcutaneously on days 1, 4, 8, and 11; and dexamethasone 40 mg orally once weekly every 21 days. Daratumumab 16 mg/kg IV was given on days 1, 8, and 15 of cycles 1 to 4 and day 1 of cycles 5 and 6. During maintenance (cycles 7-32), the patients received lenalidomide 10 mg (15 mg in cycles 10+, if tolerated) orally on days 1 to 21 every 28 days, plus daratumumab 16 mg/kg IV every 8 weeks (or every 4 weeks per patient decision after protocol amendment 2).
The primary objective of the safety run-in phase was to assess DLTs. Additional end points included safety (during mobilization and ASCT, safety data focused primarily on stem cell collection and neutrophil and platelet engraftment), the sCR rate by the end of consolidation and maintenance per International Myeloma Working Group criteria by computer algorithm,14,15 progression-free survival per International Myeloma Working Group criteria, and minimal residual disease (MRD) negativity (10−5 and 10−6 thresholds) assessed by next-generation sequencing (clonoSEQ Assay 2.0; Adaptive Biotechnologies) in the intent-to-treat population.16
The protocol and amendments were approved by the institutional review board or independent ethics committee at each site. All patients gave written informed consent. The study was conducted per the International Conference on Harmonization’s Good Clinical Practice Guidelines, the principles originating from the Declaration of Helsinki, and study site–specific regulations.
Results and discussion
Patients and treatment
Sixteen patients enrolled in the safety run-in and received D-RVd. Patient demographic and baseline characteristics are presented in Table 1. The median age was 62.5 years; 8 (50.0%) patients were male, 4 (25.0%) had International Staging System stage II or III disease, and 4 (25.0%) had high-risk cytogenetics (all del[17p]) based on the results of fluorescence in situ hybridization. All patients completed induction therapy, stem cell mobilization, ASCT, and consolidation and entered maintenance therapy. Fourteen (87.5%) patients completed the study therapy, and 2 (12.5%) discontinued the therapy because of progressive disease (n = 1) or adverse events (n = 1; neuralgia and thrombocytopenia), both during maintenance therapy.
Nine (56.3%) patients received plerixafor, and none received cyclophosphamide for mobilization. Median CD34+ cell yield was 8.05 × 106/kg (range, 3.5 × 106 to 17.6 × 106). A median of 4.72 × 106/kg (range, 2.2 × 106 to 6.0 × 106) CD34+ cells were transplanted. The median time to neutrophil (0.5 × 109/L; n = 15) and platelet (20 × 109/L; n = 16) engraftment was 14 and 13.5 days, respectively.
Safety
Three of 16 patients developed a total of 4 DLTs (fatigue, gastroenteritis, hypotension, and pneumonitis) during cycle 1; all DLTs were grade 3 and none resulted in treatment discontinuation during induction or consolidation therapy. With longer follow-up, no new safety concerns were identified. Grade 3/4 treatment-emergent adverse events (TEAEs) occurred in 15 (93.8%) patients. The most common (≥15% of patients) grade 3/4 TEAEs were neutropenia (43.8%; n = 7), pneumonia (31.3%; n = 5), lymphopenia (31.3%; n = 5), thrombocytopenia (25.0%; n = 4), and hypertension (18.8%; n = 3). One patient had TEAEs leading to discontinuation of lenalidomide and bortezomib. No grade 5 TEAEs occurred. Eleven (68.8%) patients experienced a serious adverse event. Any-grade and grade 3/4 infections occurred in 14 (87.5%) and 5 (31.3%) patients, respectively. During the maintenance phase, 5 (31.3%) patients experienced any-grade infections (the most common being upper respiratory tract infection) and 1 (6.3%) patient experienced 2 grade 3/4 infections (pneumonia and bronchitis). Infusion-related reactions occurred in 5 (31.3%) patients, and were all grade 1/2 (pruritus, chills, flushing, maculopapular rash, and vascular access site swelling). All infusion-related reactions, except for swelling of the vascular access site, occurred during the first cycle.
Efficacy
By the end of induction, all patients had responded, with a best outcome of complete response (CR) for 2 (12.5%) patients (Figure 1). By the end of D-RVd consolidation, a best response of sCR had occurred in 9 (56.3%) patients. At the last follow-up (after D-R maintenance therapy), 15 (93.8%) patients had achieved a best response of sCR. Median time to first response was 0.77 months (range, 0.1-2.1), and the median duration of response was not estimable. Median time to ≥CR was 7.36 months (range, 2.8-18.5); median duration of ≥CR was not estimable. MRD negativity (10‒5) at the end of induction occurred in 3 (18.8%) patients; none were MRD negative at 10–6. By the end of consolidation, 8 (50.0%) patients were MRD negative at 10‒5, and none were MRD negative at 10−6. At the last follow-up, 13 (81.3%) patients had achieved MRD negativity at 10‒5 and 5 (31.3%) patients at 10‒6. MRD negativity (10‒ 5) was sustained for ≥12 months in 8 (50.0%) patients.
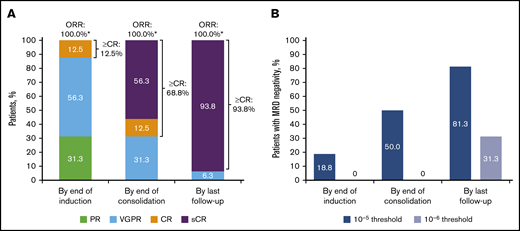
Summary of updated response rates and MRD-negativity rates (10‒5or 10−6threshold) over time for D-RVd in the safety run-in cohort (N = 16) of GRIFFIN. (A) Response rates over time are shown for the response-evaluable population (n = 16). (B) MRD-negativity rates over time are shown for the intent-to-treat population (N = 16). CR, complete response; ORR, overall response rate; PR, partial response; VGPR, very good partial response. *Percentages do not add up to 100% because of rounding.
Summary of updated response rates and MRD-negativity rates (10‒5or 10−6threshold) over time for D-RVd in the safety run-in cohort (N = 16) of GRIFFIN. (A) Response rates over time are shown for the response-evaluable population (n = 16). (B) MRD-negativity rates over time are shown for the intent-to-treat population (N = 16). CR, complete response; ORR, overall response rate; PR, partial response; VGPR, very good partial response. *Percentages do not add up to 100% because of rounding.
At a median follow-up of 40.8 months (range, 20.6-43.0), disease had progressed in 3 patients. The estimated 24- and 36-month progression-free survival rates were 93.8% and 78.1%, respectively, and the estimated 24- and 36-month overall survival rates were both 93.8%. One death occurred in the single patient who did not achieve sCR and progressed at 12.5 months.
Conclusion
With >3 years of follow-up in the safety run-in cohort of GRIFFIN, D-RVd induction, ASCT, and D-RVd consolidation led to durable hematologic responses and MRD negativity in transplant-eligible NDMM that further deepened with 2 years of D-R maintenance. These results are consistent with the deep responses seen in the randomized phase of the GRIFFIN study13 and suggest that responses in the randomized cohort will be durable and continue to improve with maintenance.
The safety profile was consistent with the known safety profiles of daratumumab and RVd, with no new safety concerns with longer follow-up. Only 1 patient discontinued treatment (lenalidomide and bortezomib), and no patients died of TEAEs. The randomized phase of GRIFFIN confirmed that D-RVd does not result in new safety concerns. In all patients, sufficient stem cells were collected for HDT and ASCT, and D-RVd did not negatively affect time to engraftment.13
These long-term data, together with the positive results of the GRIFFIN randomized phase, show that D-RVd induction with D-R maintenance is effective and well tolerated in patients with transplant-eligible NDMM. The results build on data from pivotal studies that established the clinical benefit of RVd-based therapy in this setting17 and support D-RVd as a potential new standard of care for these patients.
The data-sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Acknowledgments
The GRIFFIN team acknowledges the contributions of all principal investigators who participated in the study: Carlos Alemany, Larry D. Anderson Jr, Ashraf Badros, Ajai Chari, Robert Cornell, Luciano J. Costa, Caitlin Costello, Andrew J. Cowan, Binod Dhakal, Yvonne A. Efebera, Laura Finn, Cristina Gasparetto, Sarah A. Holstein, Andrzej Jakubowiak, Jonathan L. Kaufman, Hakan Kaya, Sarah Larson, Jacob Laubach, Tomer Mark, Nitya Nathwani, Ruben Niesvizky, Joanne Filicko-O’Hara, Robert Z. Orlowski, Brandi Reeves, Paul G. Richardson, Cesar Rodriguez, Douglas W. Sborov, Nina Shah, Kenneth H. Shain, Leyla Shune, Rebecca Silbermann, Pallawi Torka, Peter M. Voorhees, Andrew Whiteley, Tanya M. Wildes, and Jeffrey Zonder. The team also acknowledges the Alliance Multiple Myeloma Committee, particularly Phil McCarthy and our patient advocate Jim Omel, for their input and support of the study. Last, the team thanks the Alliance Foundation Trials team for their key role in the conduct of this trial (https://acknowledgments.alliancefound.org). The authors thank the patients who volunteered to participate in the trial, their families, and the staff members at the trial sites who cared for them; the members of the data review committee (Tracy McGowan [Chair], Leslie Killion, Daniela Hoehn, and Wayne Langholff) and the safety monitoring committee (Carol Ann Huff [Chair], Adam Cohen, Weichung Shih, and Parexel International, LLC [Statistical Support Group]); the representatives of the sponsor who were involved in data collection and analyses; and Grace Wang, of MedErgy, for medical writing and editorial support funded by the sponsor.
This trial was supported by research funding from Alliance Foundation Trials and Janssen Oncology.
Authorship
Contribution: P.M.V. and P.G.R. contributed to the study design, data acquisition, and data analysis or interpretation; C.R. and L.J.C. contributed to data acquisition and data analysis or interpretation; P.B., D.H., J.U., M.Q., and T.S.L. contributed to study design and data analysis or interpretation; Y.L. and H.P. contributed to data analysis or interpretation; B.R. and N.N. contributed to data acquisition; and all authors reviewed the manuscript, approved the final version, decided to publish this report, and vouch for the accuracy and completeness of the data.
Conflict-of-interest disclosure: P.M.V. has served on advisory boards for Adaptive Biotechnologies, Bristol Myers Squibb, Janssen, and TeneoBio; has served in a consultancy role for Novartis and Oncopeptides; and has received honoraria from GlaxoSmithKline and Oncopeptides. C.R. has served in a consultancy role or on the speakers’ bureaus for Bristol Myers Squibb, Takeda, and Amgen. B.R. has served on the speakers’ bureau for Bristol Myers Squibb and has received honoraria from Incyte and Takeda. L.J.C. has received research funding from Janssen and Amgen; has received honoraria from Amgen, Celgene, Sanofi, Janssen, and Bristol Myers Squibb; and has served in a consultancy role for Janssen, Amgen, Sanofi, AbbVie, Celgene, Bristol Myers Squibb, and Genentech. Y.L. and P.B. are employees of Janssen. D.H. is an employee of Merck and was an employee of Janssen at the time of the study. J.U. is an employee of Genmab and was an employee of Janssen at the time of the study. H.P., M.Q., and T.S.L. are employees of Janssen and have equity ownership in Johnson & Johnson. P.G.R. has received research funding from Oncopeptides, Celgene, Takeda, and Bristol Myers Squibb; has received honoraria from Janssen, Takeda, Celgene, GlaxoSmithKline, and Secura Bio; has served in a consultancy role for Celgene/Bristol Myers Squibb, Janssen, Karyopharm, Oncopeptides, Sanofi, Secura Bio, Takeda, AbbVie, and GlaxoSmithKline; and has served on the board of directors or held membership on an advisory committee for Jazz Pharmaceuticals. N.N. declares no competing financial interests.
The current affiliation for D.H. is Merck & Co., Kenilworth, NJ.
The current affiliation for J.U. is Genmab, Princeton, NJ.
Correspondence: Peter M. Voorhees, Levine Cancer Institute, Atrium Health, 1021 Morehead Medical Dr, Charlotte, NC 28204; e-mail: peter.voorhees@atriumhealth.org.


