

Key Points
Augmenting early GVL response by prophylactic type 1 IFN may reduce the rates of leukemic relapse after HCT in very high–risk AML.
Reciprocal toxicities, including acute GVHD and nonrelapse mortality, were not increased after type 1 IFN treatment.

Abstract
A potent graft-versus-leukemia (GVL) response is crucial in preventing relapse, the major impediment to successful allogeneic hematopoietic cell transplantation (HCT). In preclinical studies, type 1 interferon (IFN-α) enhanced cross-presentation of leukemia-specific antigens by CD8α dendritic cells (DCs) and amplified GVL. This observation was translated into a proof-of-concept phase 1/2 clinical trial with long-acting IFN-α (pegylated IFN-α [pegIFNα]) in patients undergoing HCT for high-risk acute myeloid leukemia (AML). Patients with treatment-resistant AML not in remission or those with poor-risk leukemia were administered 4 dosages of pegIFNα every 14 days beginning at day −1 before HCT. Dose selection was established by adaptive design that continuously assessed the probability of dose-limiting toxicities throughout the trial. Efficacy was evaluated by determining the 6-month incidence of relapse at the maximum tolerated dose (MTD). Thirty-six patients (median age, 60 years) received pegIFNα treatment. Grade 3 or greater severe adverse events occurred in 25% of patients, establishing 180 μg as the MTD. In phase 2, the incidence of relapse was 39% at 6 months, which was sustained through 1-year post-HCT. The incidence of transplant-related mortality was 13%, and severe grade III-IV acute graft-versus-host disease (GVHD) occurred in 11%. Paired blood samples from donors and recipients after HCT revealed elevated levels of type 1 IFN with cellular response, the persistence of cross-presenting DCs, and circulating leukemia antigen-specific T cells. These data suggest that prophylactic administration of pegIFNα is feasible in the peri-HCT period. In high-risk AML, increased toxicity was not observed with preliminary evidence for reduction in leukemia relapse after HCT. This trial was registered at www.clinicaltrials.gov as #NCT02328755.
Introduction
Graft-versus-leukemia (GVL) responses underlie the curative potential of allogeneic hematopoietic cell transplantation (HCT). Despite this effect, relapse is the most frequent cause of mortality after HCT in acute myeloid leukemia (AML).1 Increasing HCT conditioning intensity or administering donor lymphocyte infusions or maintenance chemotherapies (5-azacitidine) have not improved leukemia-free survival (LFS) as a result of the limited impacts on relapse or additional toxicity.2-5 Given the shared immunobiology, enhancing GVL is often associated with graft-versus-host disease (GVHD)-related mortality.
GVL requires T cells to respond to host alloantigens and leukemia-specific antigens.6,7 Dendritic cells (DCs) are necessary for priming T cells against viruses or tumors.8 One mechanism, termed cross-presentation, involves presenting exogenous tumor antigens on MHC class I. In humans, BDCA-3+ (CD141+) DCs specialize in cross-presentation that promotes the development of antigen-specific CD8+ T cells.9,10 Preclinical studies demonstrated that murine CD8α DCs (BDCA-3+ homologs) generate leukemia-specific T cells and GVL in vivo without aggravating GVHD.11 Cross-presentation on CD8α DCs can be enhanced by Toll-like receptor 3 agonists (polyinosinic-polycytidylic acid) or by type 1 interferons (IFNs).12,13 Type 1 IFNs also support CD8+ T-cell function by expansion of effector and memory populations.13-15 Pegylated IFN-α (pegIFNα), a long-acting formulation with greater bioavailability relative to first-generation preparations, is commercially available for the treatment of hepatitis B/C and myeloproliferative neoplasms16-18 ; however, its role in preventing relapse is not known. The full-text version of this article contains a data supplement.
Patients undergoing HCT with refractory AML often attain remission, but ∼60% will relapse within 6 months.19-23 This suggests that leukemia relapse may be reduced by augmenting early GVL. In this study, we hypothesized that enhancing GVL with pegIFNα will reduce relapse without increasing the severity of GVHD. We conducted a phase I/II clinical trial to evaluate the safety and efficacy of pegIFNα in high-risk AML (ie, principally patients not in remission at HCT). We found that early administration of type 1 IFN may limit relapse after HCT without increasing toxicity or rates of severe acute GVHD.
Methods
Patients and donors
Patients.
A phase 1/2 single-center open-label prospective clinical trial of type 1 IFN was performed in patients with high-risk AML (clinicaltrials.gov #NCT02328755). Study procedures involved screening all patients with high-risk AML proceeding to HCT at our center for eligibility from December of 2014 to January of 2019. Reasons for not enrolling included declining participation (n = 8) or not open to accrual (n = 5). Patients who did not participate (n = 13) make up the comparison cohort that is described in the “Discussion.” Eligibility consisted of 1 of the following criteria at the time of HCT: (1) a persistent leukemia defined as ≥5% myeloblasts on aspirate, or if <5%, the presence of leukemia by flow cytometry or cytogenetics (97% of enrollments) or (2) leukemia in morphologic remission with preceding evidence of very poor–risk cytogenetic/molecular features defined as complex karyotype (≥4 clonal abnormalities), monosomal karyotype, inv(3), t(3;3), t(6;9), or FLT3-ITD mutation.24-26 AML must have been resistant to 2 sequential rounds of induction chemotherapy, or if relapsed, 1 reinduction. The minimum age of enrollment was 18 years, with no maximum age. An eligibility amendment was approved to allow 1 pediatric patient (17 years old) to participate. Patients were required to have a minimum Karnofsky performance status of 70% and meet institutional requirements for organ function. This study (HUM0093471) was approved by the Michigan Institutional Review Board. All patients provided informed consent.
GVHD prophylaxis, conditioning, and supportive care.
Tacrolimus was initiated, IV, at day −3 combined with mini-dose methotrexate for matched unrelated donors. Patients were maintained on therapeutic levels of tacrolimus (5-15 ng/mL) for ≥90 days after HCT, unless there was evidence of toxicity or relapse. T-cell–depleting sera (eg, ATG) were prohibited for recipients of HLA-matched HCT. Two recipients of HLA-mismatched (haploidentical) donors received posttransplant cyclophosphamide on days 3 and 4, as well as tacrolimus and mycophenolate mofetil beginning at day 5. All conditioning was required to be myeloablative per investigator, which included clofarabine (40 mg/m2 on days −5 to −2) with busulfan (3.2 mg/kg on days −5 to −2), fludarabine (40 mg/m2 on days −5 to −2) with busulfan (3.2 mg/kg on days −5 to −2), or fludarabine (30 mg/m2 on days −7 to −5) with total body irradiation (200 cGy twice daily on days −4 to −2) for patients receiving cells from haploidentical donors (n = 2). No maintenance therapy was permitted, including donor lymphocyte infusions or chemotherapy. After completing study treatment, 2 patients with FLT3-ITD mutations received sorafenib, according to institutional standards of care. Supportive care was according to clinical practice guidelines at the University of Michigan.
Donor selection and post-HCT chimerism analysis.
Peripheral blood or bone marrow products were selected from the best available matched unrelated, matched related, or first-degree haploidentical donors. High-resolution matching was performed at HLA-A, HLA-B, HLA-C, and HLA-DRB1. For chimerism studies, peripheral blood was analyzed post-HCT for donor and recipient microsatellite markers by multiplex polymerase chain reaction and differential fluorescence. Full chimerism required establishment of ≥95% donor cells after HCT.
Study design
Treatment.
PegIFNα (PEGASYS; Genentech) was administered subcutaneously on day −1 before HCT, as well as on days 14, 28, and 42 (±7 days). Treatment was temporarily held for grade ≥ II acute GVHD and was discontinued for grades III to IV.
Phase 1.
The primary end point was to determine a maximum tolerated dose (MTD) of pegIFNα for phase 2. Three flat doses of pegIFNα were evaluated: 45, 90, and 180 μg. The trial was initiated at 90 μg using the Bayesian modified toxicity probability interval design to allow continuous assessment and adjustment of dosage throughout the trial based on the occurrence of dose-limiting toxicities (DLTs).27
Phase 2.
The primary efficacy end point was the cumulative incidence of relapse at 6 months post-HCT.
Assessment of AML response.
Remission was first assessed at day 30 (±7 days) after HCT. Complete remission (CR) status was established by achieving all of the following criteria: bone marrow myeloblasts <5% without Auer rods, absence of circulating blasts, flow cytometry without AML (sensitivity ∼0.01%), and lack of cytogenetic abnormalities. Where applicable, molecular analyses and assessment of extramedullary AML were incorporated into response assessments before and after HCT. Because assessments were performed in the early post-HCT period, full hematologic recovery was not a requirement for CR. Patients had to remain negative on all subsequent bone marrow assessments, including day 180, to be considered in CR for the primary end point. Detection of AML by any method after HCT was treated as relapse (treatment failure).
Acute GVHD assessments.
GVHD was monitored at least weekly through day 100 after HCT and subsequently at regular clinical visits through day 180 using modified Glucksberg (Keystone) consensus criteria.28
Statistics and trial end points
The estimated sample size was 35 subjects. This was based on probability estimates that assumed ≥30 subjects would be treated at the MTD. For the primary relapse end point, ≥30 patients would provide 80% power, assuming a type 1 error rate of 0.05, to show a difference between a promising 6-month incidence of 40% vs a historical incidence of 60%.19-23 The cumulative incidence for relapse was estimated using a proportional hazards model for the competing risk of nonrelapse mortality (NRM).29 The cumulative incidence of NRM and GVHD was calculated using a proportional hazards model. Overall survival (OS) and leukemia-free survival (LFS) were estimated using Kaplan-Meier methods. OS was recorded from HCT (day 0) until death, whereas LFS was recorded from day 0 until death or relapse. Replacement enrollments were allowed for patients experiencing DLTs who received ≤1 dose of pegIFNα. All analyses were performed using R (Vienna, Austria).
Correlative studies
Recipient plasma and peripheral blood mononuclear cells (PBMCs) were collected and cryopreserved prior to conditioning, as well as on days 28, 56, 100, and 180 post-HCT. After obtaining informed consent, a subset of donors had an aliquot of the stem cell product cryopreserved. Plasma cytokines and GVHD biomarkers were analyzed using Luminex (Invitrogen). Phosphorylated STAT1 (pSTAT1) staining was assessed in CD45+ PBMCs. We defined cross-presenting DC subsets as CD45+CD141+CLEC9A+ populations.30 Leukemia antigen-specific T cells were analyzed in HLA-A*0201+ patients using major histocompatibility complex (MHC) class I dextramers for Wilms’ tumor 1 (WT1; Immunodex). Leukemia-specific T cells were defined as WT1+CD8+ T cells.31 Additional details are provided in supplemental Methods.
Results
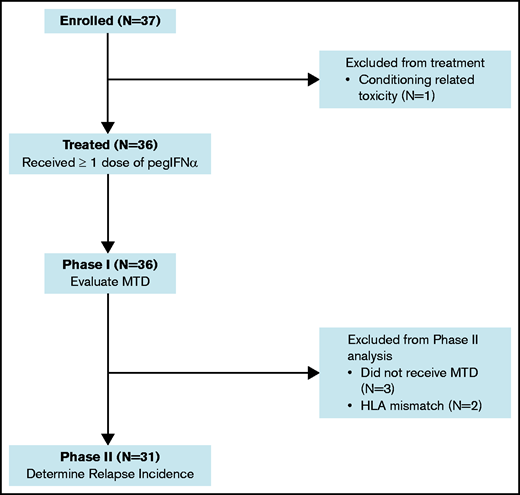
Patient characteristics and dose determination
A flow diagram for the phase 1 and phase 2 components of the study is shown in Figure 1. Thirty-six patients were treated with pegIFNα. The median age was 60 years (range, 17-72), with 64% having a hematopoietic cell transplantation-comorbidity index (HCT-CI) ≥ 3. Ninety-seven percent had detectible AML on pre-HCT disease assessments. The median disease risk score by Duval for AML not in remission was 3.32 The remaining patient characteristics are provided in Table 1.
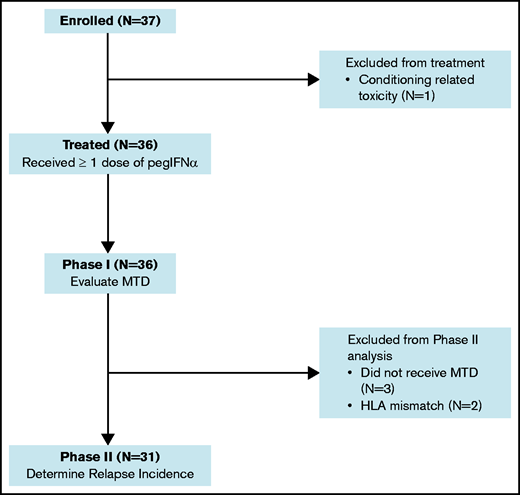
No DLT occurred in the first 3 patients treated at 90 μg; therefore, the dosage was increased to 180 μg. An engraftment failure was observed at 180 μg in a recipient of a second allogeneic HCT meeting criteria for a DLT. However, because no additional DLTs were experienced, 180 μg was determined to be the MTD in phase 1. Overall, 87% of pegIFNα dosages were administered. The most common cause for a dosage hold was acute GVHD (7%).
Engraftment and SAEs.
Neutrophil and platelet engraftment occurred at a median of 12 and 16 days, respectively. Postengraftment analysis for lineage-specific chimerism was available in 30 patients by day 100. Twenty-nine patients (97%) established full donor chimerism in the myeloid (CD33) compartment. Grade ≥ 3 nonhematologic severe adverse events (SAEs) during pegIFNα treatment were graded according to Common Terminology Criteria for Adverse Events version 4.0 (Table 2). In total, 25% of patients experienced ≥1 SAE. The most common toxicity was rash in 11% of patients that was classified as cutaneous GVHD. Two pulmonary complications were observed during treatment. One patient developed pulmonary edema related to fluid overload and another developed pneumonitis related to respiratory syncytial virus. Both pulmonary events resolved with standard supportive treatment.
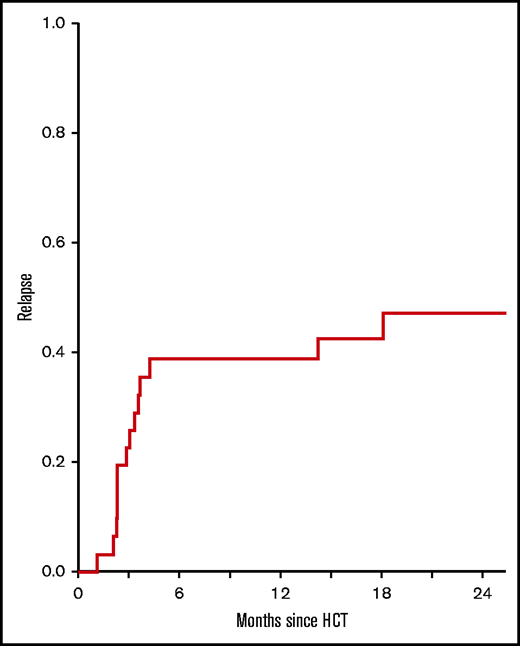
Relapse and leukemia response.
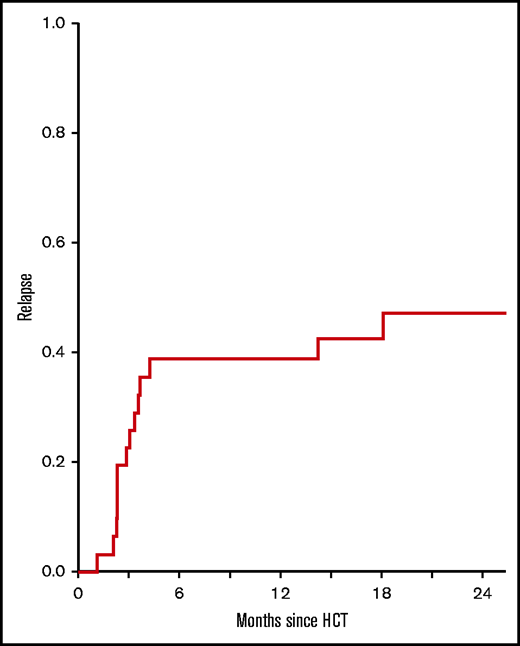
The primary end point of phase 2 was 6-month leukemia relapse in recipients of HLA-matched HCT receiving pegIFNα at the MTD (n = 31). The cumulative incidence of relapse was 39% (95% confidence interval [CI], 24-58) which was sustained at 1 year (Figure 2). For the entire study (n = 36), including phase 1 and phase 2 cohorts, the 6-month incidence of relapse was 42% (95% CI, 27-60). Upon bone marrow examination at day 30 after HCT, 34 of 36 patients (94%) had confirmed morphologic remission, including the absence of any flow cytometric, cytogenetic, extramedullary or molecular aberrations that were present prior to HCT.
NRM and GVHD.
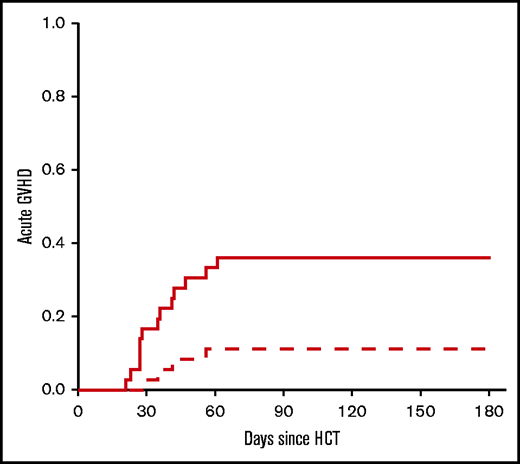
Toxicity resulting in NRM was 13% (95% CI, 5-31) and 25% (95% CI, 12-46) at 6 months and 2 years, respectively (Figure 3). Early NRM (6 months) was due to GVHD (n = 2), infection (n = 2), and graft failure (n = 1). The incidence of grade 2-4 and grade 3-4 acute GVHD was 39% (95% CI, 24-58) and 13% (95% CI, 5-31), respectively. For the entire study population, including phase 1 and phase 2, the incidence of grade 2-4 acute GVHD was 36% (95% CI, 23-54), whereas the incidence for grade 3-4 acute GVHD was 11% (95% CI, 4-27) (Figure 4). GVHD characteristics are provided in Table 3.

NRM. The cumulative incidence of NRM for patients receiving the phase 2 dosage of pegIFNα (n = 31).
NRM. The cumulative incidence of NRM for patients receiving the phase 2 dosage of pegIFNα (n = 31).
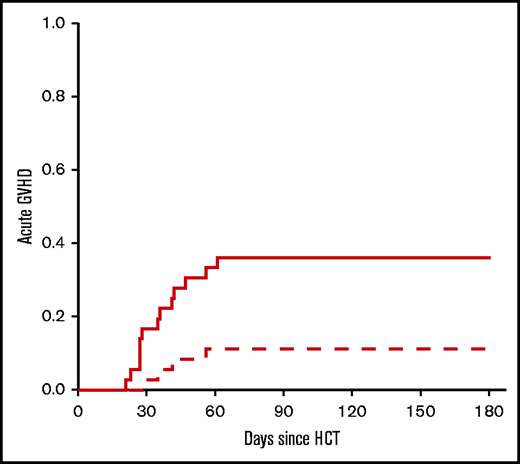
Acute GVHD. The cumulative incidence of acute GVHD by day 180 after HCT. Data include phase 1 and 2 cohorts (n = 36). Dashed line: grade 2-4; solid line: grade 3-4.
Acute GVHD. The cumulative incidence of acute GVHD by day 180 after HCT. Data include phase 1 and 2 cohorts (n = 36). Dashed line: grade 2-4; solid line: grade 3-4.
Infection.
Documented infections were recorded through day 180 by type and severity (Table 4). Overall, 72% of enrolled patients experienced ≥1 infection. Severe (grade 3) infections occurred in 9% of patients. Asymptomatic viral reactivation accounted for 53% of infections commonly involving cytomegalovirus viremia. Among patients with cytomegalovirus reactivation, 58% required antiviral treatment, and 1 patient developed viral enteritis. Bacterial infections occurred in 42% of patients, predominantly staphylococcal or enterococcal bacteremia before engraftment. There were no grade 3 bacterial infections. Complete infection data are provided in supplemental Table 1.
Survival
In phase 2, OS was 55% (95% CI, 40-75) and 33% (95% CI 19-58) at 6 months and 2 years, respectively (Figure 5A). LFS was 48% (95% CI 34-70) and 28% (95% CI, 15-52) at 6 months and 2 years, respectively (Figure 5B). For the entire study, OS was 53% (95% CI, 39-72) and 32% (95% CI, 19-54) at 6 months and 2 years, respectively. There were no differences in OS or LFS by age, conditioning therapy, donor type, cytogenetic risk, pre-HCT blast percentage, or HCT-CI.
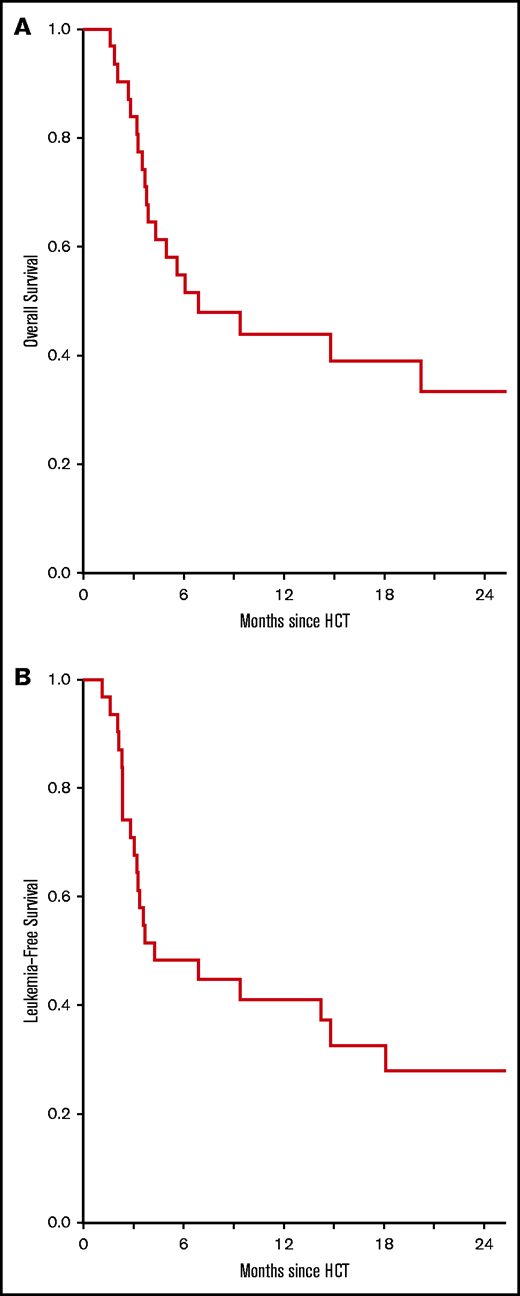
Survival. OS (A) and LFS (B) for patients receiving the phase 2 dosage of pegIFNα (n = 31).
Survival. OS (A) and LFS (B) for patients receiving the phase 2 dosage of pegIFNα (n = 31).
Correlative studies
Plasma type 1 IFN, pharmacodynamics, and inflammatory markers.
Paired plasma and PBMC samples were analyzed in a subset of patients before conditioning, as well as at days 28 and 56, reflecting time points immediately prior, during, and after completing pegIFNα. Plasma levels of IFN-α increased significantly at days 28 and 56 compared with measurements before treatment (P < .01; Figure 6A). Levels of other type 1 IFN (IFN-β) and type 2 IFN (IFN-γ) did not change (supplemental Figure 1). We then evaluated PBMCs for IFN-α signaling by pSTAT1. The median frequency of CD45+ cells expressing pSTAT1 increased at day 28 compared with baseline levels before infusion (P < .05; Figure 6B). Cytokines, cytolytic granules, and GVHD biomarkers were assessed as corresponding markers of inflammation. Plasma tumor necrosis factor-α, granzyme A, and ST-2 increased compared with baseline; however, other measured cytokines (IL-1β, IL-6, IL-15) did not (supplemental Figure 1).
![Type 1 IFN levels, pharmacodynamics response, and cellular immune subsets. (A) Paired plasma levels of IFN-α in recipients (n = 12) at baseline (preconditioning [Pre]), as well as at day 28 (D28) and day 56 (D56), were analyzed by Luminex array. (B) Frequency of pSTAT1 protein (n = 8) within CD45+ cells measured by fluorescence-activated cell sorting (FACS) in paired samples from donors and recipients at day 28 and day 56. (C) The numbers of CD141+CLEC9A+ DCs (n = 3) were measured by FACS from donors and recipients at day 28 and at day 56. (D) The numbers of WT1+CD8+ T cells by WT1-specific dextramers (n = 3) in patients with HLA-A*0201. Analysis was performed by FACS in paired donors and recipients at days 28, 56, 100, and 180 after HCT. Samples obtained after relapse or high-dose corticosteroids for GVHD were excluded. Data are mean values; error bars represent the standard error of the mean. *P < .05, **P < .01, paired Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004908/2/m_advancesadv2021004908f6.png?Expires=1768463855&Signature=Mx0JGPhQ1ft6EoBa3Y1RKmKGHk9XlOGdvkXbK~f0Fdy9DwuHnQzsyvDI2nZo6a0JnWqY96qD0Vp2wRfZS9mH~9X6-CrZpMW1qu3p2GxwsmyUJUo-xSJH5RzKzgi1EiuEx12ry0oLb4Tzj1yB2DtrWlkj-vtg5kEL-Cf042wtxSms7hnVItYBS244ih4fk1wHtn6APH00SCEqmwQrTOncrnVXW1-ZVsnVaZGs52Q0J944UGWVU5LkiIZI8oEqk96hkJU-LNE0ae0ryweCIslnAJpQs-TjaDd4JXnIoO4S7~xQsrVKRjTqYy6i62lCkX9UB2ZXUFME3GmXsCl3qF-Liw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Type 1 IFN levels, pharmacodynamics response, and cellular immune subsets. (A) Paired plasma levels of IFN-α in recipients (n = 12) at baseline (preconditioning [Pre]), as well as at day 28 (D28) and day 56 (D56), were analyzed by Luminex array. (B) Frequency of pSTAT1 protein (n = 8) within CD45+ cells measured by fluorescence-activated cell sorting (FACS) in paired samples from donors and recipients at day 28 and day 56. (C) The numbers of CD141+CLEC9A+ DCs (n = 3) were measured by FACS from donors and recipients at day 28 and at day 56. (D) The numbers of WT1+CD8+ T cells by WT1-specific dextramers (n = 3) in patients with HLA-A*0201. Analysis was performed by FACS in paired donors and recipients at days 28, 56, 100, and 180 after HCT. Samples obtained after relapse or high-dose corticosteroids for GVHD were excluded. Data are mean values; error bars represent the standard error of the mean. *P < .05, **P < .01, paired Student t test.
Type 1 IFN levels, pharmacodynamics response, and cellular immune subsets. (A) Paired plasma levels of IFN-α in recipients (n = 12) at baseline (preconditioning [Pre]), as well as at day 28 (D28) and day 56 (D56), were analyzed by Luminex array. (B) Frequency of pSTAT1 protein (n = 8) within CD45+ cells measured by fluorescence-activated cell sorting (FACS) in paired samples from donors and recipients at day 28 and day 56. (C) The numbers of CD141+CLEC9A+ DCs (n = 3) were measured by FACS from donors and recipients at day 28 and at day 56. (D) The numbers of WT1+CD8+ T cells by WT1-specific dextramers (n = 3) in patients with HLA-A*0201. Analysis was performed by FACS in paired donors and recipients at days 28, 56, 100, and 180 after HCT. Samples obtained after relapse or high-dose corticosteroids for GVHD were excluded. Data are mean values; error bars represent the standard error of the mean. *P < .05, **P < .01, paired Student t test.
DC subsets and leukemia-specific T cells.
We first confirmed whether CD141+ DCs, a rare population involved in antigen cross-presentation, were present in paired donor and recipient samples.9 CD141+CLEC9A+ DCs detected in the donor inoculum decreased in number but persisted on days 28 and 56 after HCT (Figure 6C). Having confirmed their presence, we then explored the function of cross-presentation by measuring leukemia-specific antigen CD8 T cells. We analyzed MHC class I dextramers loaded with WT1, an antigen that is commonly overexpressed in AML, in patients with HLA-A*0201 alleles. WT1+CD8+ T cells were detected at low frequencies in donors and at day 28 in recipients, but they showed a trend toward sustained persistence at day 180 (Figure 6D).
Discussion
In this phase 1/2 clinical trial, we evaluated the safety and efficacy of augmenting GVL responses after HCT. The data suggest that administration of exogenous IFN-α does not alter toxicity, as measured by rates of adverse events, acute GVHD, or NRM. Importantly, the incidence of relapse was lower than hypothesized, meeting the study’s primary end point. This suggests that type 1 IFNs are potentially effective in reducing relapse in high-risk AML and may increase OS, findings that will require confirmation in larger controlled trials.
Increasing GVL can be accomplished experimentally by enhancing cross-presentation, which enables expression of exogenous antigens within MHC class I molecules presented on DCs.11,13,14 This results in priming of CD8+ T cells toward tumors or leukemia antigens. In preclinical models, targeting cross-presentation with polyinosinic-polycytidylic acid or type 1 IFN improved LFS without aggravating GVHD.11,13 Our phase 1 data suggest that pegIFNα is safe when administered every 2 weeks beginning on day −1 before stem cell infusion. DLTs were infrequent, occurring in 1 patient (engraftment failure). The remaining patients exhibited normal hematologic engraftment, suggesting that this was an isolated event in an at-risk subject (prior allogeneic HCT). The frequency of other SAEs were generally acceptable; rash (11%) and pulmonary events (6%) were the most common. Consequently, 180 μg was selected as the phase 2 dose of pegIFNα. Nonetheless, considering the limited sample size, HCT complications will require further monitoring in larger studies.
Previous case series using short-acting type 1 IFNs for treating post-HCT relapse indicated that half of patients experience SAEs or acute GVHD.33,34 Several reasons may account for the low rate of SAEs in our study. First, our study was performed as prevention, in contrast to previous studies that administered IFN-αas treatment for relapse after HCT . Second, the treatment schedule was brief, and the dosages were moderate compared with previous studies. Third, in our study, pegIFNα was administered in conjunction with calcineurin inhibitors for GVHD prophylaxis. This contrasts with previous studies in which IFN-α treatment was combined with reduced immune suppression or additional immunotherapies (IL-2, DLI).35 Another key safety finding of our study was the 11% incidence of severe acute GVHD and 25% incidence of NRM, suggesting that pegIFNα does not significantly amplify toxicity or alloreactivity toward host tissues, similar to experimental studies. Prospective studies have reported GVHD rates of 38% to 50% and NRM of 25% to 36% in patients undergoing HCT with persistent AML,20 whereas retrospective analyses suggest NRM > 40%.36,37
To determine whether type 1 IFN is effective in reducing relapse, we selected an exceptionally high-risk population (ie, patients undergoing HCT with detectable AML). Although other factors, such as mutational status of AML, influence prognosis, undergoing HCT while not in remission is the best known predictor for relapse and mortality.38,39 At day 30 after HCT, 34 of 36 patients had confirmed CR; however, this may reflect transient leukemia clearance similar to other studies that used myeloablative regimens.20,36 Of greater importance, relapse incidence was 39% at 6 months and at 1 year suggesting that, consistent with our hypothesis, early administration of pegIFNα results in sustained remissions. Therefore, it is plausible that, as has been shown in preclinical studies, type 1 IFN promotes licensing of CD141+ DCs that are specialized in cross-presentation, thereby enhancing early GVL.6,10,11,13,40 Although our study did not distinguish effects on host vs donor DCs, it does show that CD141+ DCs present in the donor inoculum persist early after HCT. Furthermore, new epigenetic and mutational modifiers, such as FLT3 and IDH1 inhibitors, represent promising strategies for the reduction of relapse post-HCT; however, their direct effects on immune cells and immune-mediated GVL responses are understudied and will require ongoing assessments in experimental and clinical studies.
The net effect of elevated NRM and relapse is poor survival. The 2-year OS and LFS from the study were 33% and 28%; although not optimal, they are suggestive of a tangible improvement in survival compared with other HCT studies. Previous prospective and retrospective studies of refractory AML with morphologic or MRD-positive disease at the time of HCT revealed a relapse incidence of 55% to 65% after HCT, with OS ranging from 14% to 26%.20,21,23,32 Similarly, although an imperfect comparison, contemporaneous patients with AML with similar eligibility who did not participate in our study had a 2-year OS of 15% in outcome data from University of Michigan (J.M. and P.R., unpublished data; supplemental Figure 2). Thus, type 1 IFN may control relapse without reciprocal increases in toxicity and improved OS. Given that our trial involved heterogeneous donor types, variations in leukemia subtypes (≥5% vs <5% blasts, various cytogenetic risk), and a limited sample size, improvements in relapse and survival outcomes will require confirmation in a larger randomized trial.
To describe the levels of type 1 IFN during the study, paired blood samples were examined in a subset of donors and recipients. Significant elevations in plasma IFN-α were detected after HCT, along with increased pSTAT1 in PBMCs. However, without untreated controls these elevations may reflect endogenous IFN-α release that results from the inflammatory conditions of HCT. Nonetheless, recent experimental data suggest that lack of cross-presentation and T-cell exhaustion diminish GVL.40 Our trial shows that CD141+CLEC9A+ DCs contained in the donor inoculum persist after conditioning, and that leukemia-specific T cells were identified 6 months after HCT. These findings, together with lower-than-expected rates of relapse, indicate the possibility of a sustained antileukemic T-cell response. The low numbers of circulating cells, lack of controls, and small trial size make these data descriptive in nature, thus precluding any direct correlation with pegIFNα dosing or clinical response. Furthermore, because most immune cells express type 1 IFN receptors, our study did not distinguish between cross-presentation and direct effects on other cell types (natural killer cells). Well-controlled clinical studies will facilitate further assessment of the key immunologic targets of type 1 IFN in humans.
In summary, we provide evidence that exogenous type 1 IFN is a potentially safe and feasible strategy to limit post-HCT relapse in high-risk AML. The use of long-acting IFN-α for prevention may augment the antileukemia response and potentially improve OS. These data require validation in a prospective randomized trial.
Acknowledgments
The authors are solely responsible for the design, data collection, analysis, and decision to publish this trial.
P.R. is supported by National Institutes of Health National Cancer Institute grants R01 CA203542 and R01 CA217156 and National Institutes of Health National Heart, Lung, and Blood Institute grants R01 HL152605 and P01 HL149633. J.M.M. is the recipient of a National Institutes of Health Career Development Award (K23 AI123595) and a Rogel Cancer Center Scholarship. The Elsa U. Pardee Foundation provided funding for research and drugs (pegIFNα).
Authorship
Contribution: J.M.M. and P.R. designed the study; J.M.M., A.P., M.R., S.A., M.G., M.T., J.M., B.P., G.Y., S.W.C., and P.R. recruited patients and collected, assembled, analyzed, and interpreted clinical data; D.P., J.M.M., B.P., and P.R. designed, performed, and/or analyzed correlative data for immune subsets; T.B. performed statistical analyses and contributed to the statistical design of the trial; and all authors wrote, reviewed, and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John M. Magenau, Blood and Marrow Transplant Program, Division of Hematology-Oncology, University of Michigan, 1500 E. Medical Center Dr, SPC 5948, Ann Arbor, MI 48109-5948; e-mail: johnmage@med.umich.edu; and Pavan Reddy, Blood and Marrow Transplant Program, Division of Hematology-Oncology, University of Michigan, 1500 E. Medical Center Dr, SPC 5948, Ann Arbor, MI 48109-5948; e-mail: reddypr@med.umich.edu.




![Type 1 IFN levels, pharmacodynamics response, and cellular immune subsets. (A) Paired plasma levels of IFN-α in recipients (n = 12) at baseline (preconditioning [Pre]), as well as at day 28 (D28) and day 56 (D56), were analyzed by Luminex array. (B) Frequency of pSTAT1 protein (n = 8) within CD45+ cells measured by fluorescence-activated cell sorting (FACS) in paired samples from donors and recipients at day 28 and day 56. (C) The numbers of CD141+CLEC9A+ DCs (n = 3) were measured by FACS from donors and recipients at day 28 and at day 56. (D) The numbers of WT1+CD8+ T cells by WT1-specific dextramers (n = 3) in patients with HLA-A*0201. Analysis was performed by FACS in paired donors and recipients at days 28, 56, 100, and 180 after HCT. Samples obtained after relapse or high-dose corticosteroids for GVHD were excluded. Data are mean values; error bars represent the standard error of the mean. *P < .05, **P < .01, paired Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004908/2/m_advancesadv2021004908f6.png?Expires=1768464102&Signature=kitebYRDWv1k4Zy3ibKfd0oDukDewMkd1a8QSKhCsu4xjoh-5UIcOv~vzeS5IJVpzqlS-fGf3jVPXrJRZUu2OUYiB-adxQzFFF5UqcqJP0x3bFM1c0vLfSc-R7k9gXGM81Ssj7gCai71X~aqy7zY9L5SQtsjJgyqKZQSamFzk-V7QJCBgen6FmF02AhsWTagnfmy7YAnlG0R5PMr8UDbBGBqjEowIbBJlget3SilFcikuNrWagvuKpsAwlegPSCt6-gwDH6p6d5dwIJl2QRgA5E0YoP8dSOgB-TRjIoAUjt8LQeeZAmtS8-l7o3vhDETk30XlSqBBPuQwFj4N2EIfQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)

