

TO THE EDITOR:
Red blood cell (RBC) clearance occurs principally in the spleen and liver,1 although the precise mechanisms remain unclear. In sinusoidal spleens, as in humans, rats, and dogs, attention has focused on the interendothelial slits of the red pulp where passage may become difficult because of reduced deformability due to ageing or disease states (eg, sickle cell disease2 or malaria3,4 ). However, even if blocked in the red pulp, the question of how RBCs are finally removed remains open. This same question is of paramount importance for clearance in the spleens of mice, horses, cats, or even zebrafish, whose spleens are nonsinusoidal.5 Various mechanisms have been proposed that include, among other things, exposure of phosphatidylserine,1,6 conformational changes in protein CD47,7 oxidation of proteins (eg, band 3),8 reduction in membrane or whole cell deformability,9 and, importantly for what follows, expression of adhesion molecules.10 However, in all of this, a complete mechanistic pathway for clearance remains undefined.
Klei et al11 recently proposed that the pathway to RBC clearance involves cell adherence to the reticular meshwork of the red pulp or to the sinuses, while under shear flow, that leads to hemolysis and the formation of ghost cells; their hypothesis envisions that ghost cells, and not intact red cells, are ingested by red pulp macrophages. This schema may indeed provide a unifying concept that carries over for RBC clearance in a wide range of species. Klei et al11 support their hypothesis by demonstrating that only a low percentage of intact RBCs are ingested by red pulp macrophages of the human spleen and that hemolysis and transition to ghost cells occur in senescent-like RBCs when subjected to sustained shear flow.
At the same time, Asaro et al12 published a computational analysis of adhered RBCs in shear flow that concluded that senescent-like RBCs (ie, those with compromised skeletal-membrane connectivity) tend to undergo vesiculation while adhered to the vascular environment under shear flow, leading to perhaps evagination and lysis. An important feature of their analysis was the time scales of adhesion under shear flow because they found, and predicted, that sufficient time was required for skeletal remodeling to reduce skeletal-membrane cohesion to induce vesiculation and possible eventual lysis. They argued that this skeletal remodeling also involved the time-dependent formation of localized skeletal-membrane separations (ie, “imperfections”) that lead to membrane blebs and vesiculation. Common definitions of red cell deformability rarely consider such time-dependent processes. Thus, their analysis provides a mechanistic description of why and how the processes of lysis and ghost formation reported by Klei et al11 occur. They further argued that such processes could and would occur more generally within the vasculature and, especially, the microvasculature and may contribute to the progression of various disease states, such as atherosclerosis and retinal vein occlusion. Hence, it is worthwhile, if not necessary, to look more closely at the full details of these common scenarios and imagine the holistic pathway from a compromised RBC-to-adherence under sustained shear flow-to-vesiculation-to-hemolysis-to-ghost cell-to-clearance. This pathway defines a specific sequence of biochemical mechanisms that lead to skeletal-membrane “imperfections” and the time dependence of their formation.
Figure 1A-C shows results for an RBC adhered to a substrate and subjected to a shear flow stress of τ = 0.7 dynes/cm2. Note that this number compares well with the value used by Klei et al11 of τ = 0.5 dynes/cm2 in demonstrating hemolysis and ghost formation of an adhered RBC (see their Figure 4B11 ). This simulation was patterned directly from the videomicroscopy of red cells adhered to the red pulp and sinuses of rat spleens5 ; the deformation patterns in Figure 1A and in Groom et al5 are nearly identical. Figure 1B illustrates the area deformation of the RBC skeleton as it expands and as transmembrane protein attachments (ie, pinning points) are dragged laterally. Pinning points involve mobile transmembrane proteins and are indicated in Figure 1D as junctional complexes (JCs) or suspension complexes.14 The area deformation of protein 4.3, as shown, represents a nearly 80% reduction in the area density of skeletal pinning points. This, by itself, represents a reduction in the “cohesive strength” of the skeleton-to-membrane. Figure 1C shows the contact pressure between the skeleton and membrane that develops as a result of the difference in tensions in each (ie, tension in the skeleton [Ts] and tension in the membrane [Tm] that are indicated in Figure 1D). As seen, the contact pressure becomes quite large in the tip area and negative; the value of 160 Pa is of a magnitude that would tend to promote membrane-skeleton separation.14 Their further analysis13 of vesiculation demonstrated that, under these conditions, when such decohesion events occur locally they lead to spontaneous vesiculation; here again the question of the time scale for such reactions becomes vital.
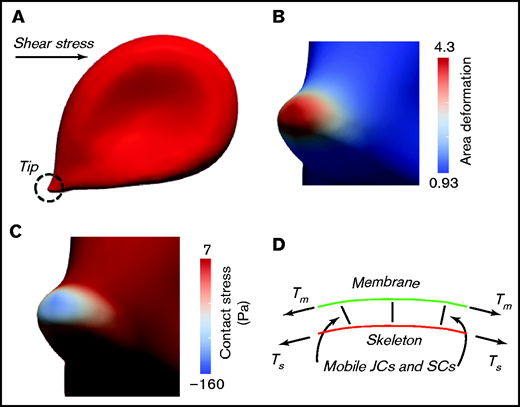
Mechanical state of an adhered RBC in shear flow. (A) An RBC locally adhered while under a shear stress of τ = 0.7 dynes/cm2. Note the encircled “adhered tip.” (B) Area deformation of the skeleton, at the adhered tip, as measured by λ1, λ2, where λi, with i = 1,2 are the 2 principal stretches in the plane of the membrane; this means that the fractional area change of the skeleton would be α = λ1λ2 − 1. Note that the area deformation of the bilipid membrane is 0. (C) The contact pressure at the adhered tip, which when negative, represents a stress that tends to pull apart the skeleton and membrane. (D) The model is that of a viscous bilipid membrane attached to the skeleton via discrete “pinning points” representing the transmembrane protein and peripheral anchoring proteins. Note that the membrane proteins are mobile within the membrane via viscous drag; hence, the skeleton may “remodel” and, thereby, alter the areal density of pinning points. Tm and Ts are the tensions in the membrane and skeleton, respectively. Details can be found in Asaro et al.12,13 SC, suspension complex.
Mechanical state of an adhered RBC in shear flow. (A) An RBC locally adhered while under a shear stress of τ = 0.7 dynes/cm2. Note the encircled “adhered tip.” (B) Area deformation of the skeleton, at the adhered tip, as measured by λ1, λ2, where λi, with i = 1,2 are the 2 principal stretches in the plane of the membrane; this means that the fractional area change of the skeleton would be α = λ1λ2 − 1. Note that the area deformation of the bilipid membrane is 0. (C) The contact pressure at the adhered tip, which when negative, represents a stress that tends to pull apart the skeleton and membrane. (D) The model is that of a viscous bilipid membrane attached to the skeleton via discrete “pinning points” representing the transmembrane protein and peripheral anchoring proteins. Note that the membrane proteins are mobile within the membrane via viscous drag; hence, the skeleton may “remodel” and, thereby, alter the areal density of pinning points. Tm and Ts are the tensions in the membrane and skeleton, respectively. Details can be found in Asaro et al.12,13 SC, suspension complex.
We next envisioned that, with a red cell adhered under shear flow, an influx of Ca++ occurs through Piezo1, as shown in Figure 2A. Influx through Piezo1 activates the Gardos channel,15,16 leading to an efflux of K+ and H2O and initial dehydration of the cell. However, with time, Ca++ will activate protein kinase C (PKC), which leads to near-term binding of calmodulin (CaM) to protein 4.1R17,18 and decreased membrane stability that is due to decreases in 4.1R-stimulated bonding of spectrin to actin19 ; slightly later, activation of PKC induces phosphorylation of protein 4.1R.20,21 Evidence suggests that Ca++/CaM reduces the affinities of the spectrin-actin-adducin and spectrin-actin-4.1R interactions, both of which occur at the JC.18 The phosphorylation of 4.1R, also at the JC, disrupts the association between spectrin and actin.20,21 The time scale for CaM-induced disruptions is of order O(1-3 minutes), whereas that caused by PKC via protein 4.1R is of order O(5-90 minutes). Both effects induce a weakening of the skeleton-membrane connection that augments the reduction in the areal density of skeleton attachments depicted in Figure 1D; coupled with the elevated contact pressure of Figure 1C, this leads to expected vesiculation, as depicted in Figure 2B.13 Under the schema described above, we might expect vesiculation to begin in a matter of only 5 to 10 minutes, but most it likely continue for hours.
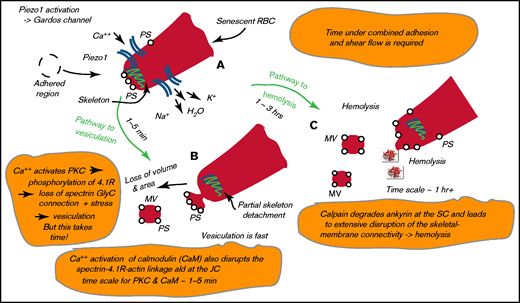
Pathway to hemolysis. (A) High membrane tension activates Piezo1, and the uptake of Ca++ activates the Gardos channel. (B) Ca++ activation of CaM and PKC leads to disruption of the skeleton-membrane connectivity. (C) Calpain degrades ankyrin, causing skeleton-membrane disruption. PS, phosphatidylserine; SC, suspension complex.
Pathway to hemolysis. (A) High membrane tension activates Piezo1, and the uptake of Ca++ activates the Gardos channel. (B) Ca++ activation of CaM and PKC leads to disruption of the skeleton-membrane connectivity. (C) Calpain degrades ankyrin, causing skeleton-membrane disruption. PS, phosphatidylserine; SC, suspension complex.
For additional insight, we next note the results of Cueff et al22 that were collected at different times after Ca++ loading using the Ca++ ionophore A23187. Their osmotic fragility curves (OFCs) show a classic short time shift left indicating dehydration, followed by a shift to the right over periods of time of order O(1-4 hours); such rightward shifts, which are usually attributed to rehydration, were reported to be due to a loss in membrane area caused by vesiculation. We also note that there was no shear stress applied in their experiments. Cueff et al22 demonstrated that the rightward OFC shifts were associated with sustained Ca++ uptake by showing the near absence of such shifts in cells dehydrated using valinomycin ionophore, a K+ transporter, or NS309, a modulator of the Gardos channel; both led to cell dehydration without Ca++ uploading.22 The time scale of the observed rightward shifts are noteworthy. Here, we recall that Klei et al11 observed the hemolysis of adhered senescent-like cells under sustained shear flow and the transition to ghost cells via hemolysis only after times of order O(2-3 hours).
We next mention the Ca++-dependent protease calpain that is known to degrade skeleton binding proteins, including ankyrin, band 3, as well as 4.1R.23,24 This disrupts the band 3-ankyrin-spectrin suspension complex and, along with the disruption at the JC, leads to a far more complete, albeit localized, disruption of the skeleton-membrane connection. The concentration of Ca++ required to activate calpain in erythrocytes is ∼10 μm.24 When Ca++ concentrations are increased, as described above, and calpain is activated, we note from Hall and Bennett24 that “calpain cleavage of ankyrin decreases the affinity for anion transporter by 8-fold and reduced binding of ankyrin would be expected to increase the lateral mobility of anion transporter in membrane.” The time scales for such protein degradation appear to be of order O(∼1 hour) or longer.25
Hemolysis would only take place after sufficient disruption of the skeleton-membrane connectivity occurred and membrane tension alone supported the full pulling force on the whole cell. This would open pores in the membrane whose osmotic fragility was already increased, as evidenced by the rightward shifts in the OFCs of Cueff et al.22 Figure 2C is a depiction of this process. Verification of this hypothesis could lead to a unifying paradigm for red cell clearance, as well as the role(s) that RBCs play in disease progression.
Authorship
Contribution: R.J.A. performed research and wrote the manuscript and P.C. performed research and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert J. Asaro, Department of Structural Engineering, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093; e-mail: scipio394@gmail.com.


