

Key Points
Patients with CLL sequentially resistant to both BCL2 and covalent BTK inhibition have a poor prognosis and represent an area of unmet need.
Abstract
Covalent Bruton tyrosine kinase inhibitors (BTKi’s) and the B-cell lymphoma 2 (BCL2) inhibitor venetoclax have significantly improved outcomes for patients with chronic lymphocytic leukemia (CLL), especially those with biologically adverse disease. Patients with CLL resistant to their first targeted agent (TA) can be effectively treated with the alternative class. However, relapses are expected with second-line TA therapy, and the clinical challenge of double class-resistant disease is now emerging with increasing frequency. To define the characteristics and outcomes of patients with double class-resistant disease, we retrospectively analyzed 17 patients who developed progressive disease (PD) on both TA classes for CLL (venetoclax, then BTKi, n=12; BTKi, then venetoclax, n = 5). The cohort was heavily pretreated (median lines of prior therapy, 4) and enriched for adverse disease genetics (complex karyotype, 12 of 12 tested [100%]; del(17p)/TP53 mutations, 15 of 17 [88%]). The median time to progression on prior venetoclax was 24 months (range, 6-94 months) and was 25 months (range, 1-55 months) on prior BTKi. Progression on second-line TA was manifest as progressive CLL in 11 patients and as Richter transformation in 6. The median overall survival after progression on second-line TA was 3.6 months (95% confidence interval, 2-11 months). Patients with double class-resistant CLL have a dismal prognosis, representing a group of high unmet need.
Introduction
Highly active targeted agents (TAs), specifically covalent Bruton tyrosine kinase (BTK) inhibitors (BTKi’s) and the B-cell lymphoma 2 (BCL2) inhibitor venetoclax, have transformed the treatment of chronic lymphocytic leukemia (CLL), particularly for patients with high-risk disease biology.1-4 Nevertheless, progressive disease (PD) on TAs is frequently observed with longer-term follow-up of patients pretreated with chemoimmunotherapy, especially in the context of TP53 dysfunction and genomic complexity.4-8 BTKi resistance is typically mediated by BTK or phospholipase C gamma 2 (PLCγ2) mutations,9 whereas venetoclax resistance is characterized by heterogenous mechanisms including BCL2 mutations conferring reduced venetoclax binding,10,11 overexpression of alternative prosurvival proteins,10,12 metabolic reprogramming,13 and others.14 Consistent with these distinct resistance mechanisms, patients with PD after their first TA frequently achieve disease control with the alternative class.15-17 However, as more patients are sequentially treated with venetoclax and BTKi’s, patients with double class-resistant disease and limited treatment options will be encountered with increasing frequency. Here, we report the clinicopathological features, management, and outcomes of patients with dual covalent BTKi- and venetoclax-resistant CLL.
Methods
Study patients
We retrospectively reviewed records of patients at the Royal Melbourne Hospital and Peter MacCallum Cancer Centre treated between June 2011 and November 2020 with a covalent BTKi or venetoclax for CLL (n = 165). Forty-two patients had been exposed to both classes. Nineteen patients had PD on both TA classes, although 2 patients received their second-line TA for Richter transformation (RT); therefore, 17 patients developed PD after receiving both TA classes for CLL (supplemental Figure 1A).
Clinical data
Patient and disease characteristics at progression on second-line TA were recorded, including age, prior therapies, fludarabine refractoriness (failure to respond or PD ≤6 months after fludarabine-based therapy),18 prior diagnosis and management of RT, del(17p)/TP53 mutations, immunoglobulin heavy chain variable region mutation status, and complex karyotype (CK) (≥3 clonal chromosomal abnormalities on conventional metaphase karyotyping).19,20 TP53 variants were detected with a sensitivity of 5% variant allele frequency using targeted next-generation sequencing of all coding exons of TP53 (NM_000546.5) and were assessed for pathogenicity using curation sources including the Genome Aggregation Database, the Catalogue of Somatic Mutations in Cancer, and the International Agency for Research on Cancer (IARC) TP53 Database. BCL2, BTK, and PLCγ2 mutations were detected using targeted next-generation sequencing. The best response to each TA, as assessed by clinical investigators using the 2018 International Workshop on CLL (iwCLL) criteria,20 was documented. Undetectable measurable residual disease was defined as detection of <1 CLL per 10 000 leukocytes in peripheral blood or bone marrow by multiparameter flow cytometry, analyzing >200 000 leukocytes.21 All patients included in the analysis provided written informed consent, and the studies were performed in line with the Declaration of Helsinki with approval from the local human research ethics committee (20/204L).
Statistical analysis
The Kaplan-Meier method was used to estimate overall survival (OS) after PD on second-line TAs. Patients without an event were censored at date of last follow-up or data cutoff (1 November 2020) if last follow-up occurred later. Associations between clinicopathological variables and OS were analyzed using the log-rank test with α = 0.05. Analyses were performed using STATA (version 14.1 for Mac) and GraphPad Prism (version 9.0.1 for Mac).
Results and discussion
Patient characteristics at PD on second-line TAs are summarized in Table 1. All instances of double class-resistant CLL emerged in the context of sequential TA therapy, and no patients had received combination TA therapy prior to the development of double class-resistant disease. The median age was 76 years (range, 52-92 years). All patients had received prior chemoimmunotherapy and the median number of prior therapies was 4 (range, 2-8). Ninety-four percent of patients had received prior fludarabine-cyclophosphamide-rituximab and 53% had fludarabine-refractory disease before first-line TAs. One patient had received a prior phosphatidylinositol 3-kinase (PI3K) inhibitor. The frequency of del(17p)/TP53 mutations was 88%. Disease karyotype was complex in all 12 evaluable patients (≥5 lesions in 8 cases [66%]). After a second-line TA, a new CK was detected in 3 patients who previously had no CK, and a new TP53 mutation was detected in 1 patient previously untested. At any time after TA exposure, BCL2 mutations were detected in 6 patients; BTK/PLCγ2 mutations were detected in 8 (Figure 1).
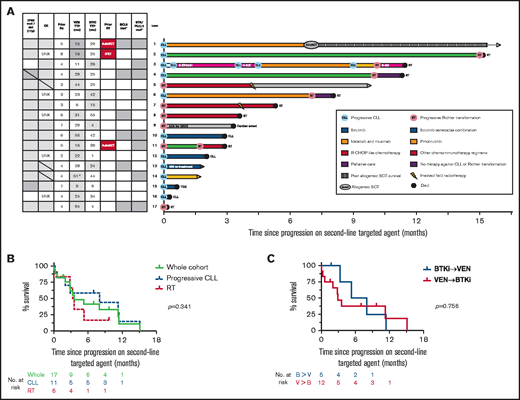
Outcomes for patients who developed PD on a second-line TA (BTKi or venetoclax). (A) Individual patient characteristics and timelines of outcomes and treatments after the development of PD on either BTKi or venetoclax used sequentially as a second-line TA. Arrows at the end of lanes indicate ongoing survival at last follow-up, circles indicate death, and other treatments have specific symbols as indicated. The columns on the left indicate clinicopathological variables at time of progression on the second-line agent. Gray fill indicates presence of a variable; gray horizontal line indicates new genetic lesion at time of progression on second-line TA; white fill indicates absence; patients who were treated for RT prior to the second-line TA are denoted by a red fill. ^The patient in lane 14 ceased venetoclax in measurable residual disease–positive CR after 6 months, resumed for progressive CLL 12 months later, then subsequently progressed while on drug with a total duration of disease control with venetoclax of 51 months. *Gray fill indicates detection of resistance mutation at any time after exposure to TA; white fill, untested or not detected. (B) OS after the development of PD on a second-line TA. Curves represent outcomes for the overall cohort (green), patients with progressive CLL on a second-line TA (blue, dashed), and patients with RT on second-line TA (red, dashed). (C) OS after the development of PD on a second-line TA, stratified by prior sequencing of TAs. Curves represent the outcomes for patients who receiving BTKi’s then venetoclax (blue) or venetoclax then BTKi’s (red). AutoSCT, autologous stem cell transplant for RT prior to second-line TA; AZA, azacitidine; B, BTKi; IBR, ibrutinib; IFRT, involved-field radiotherapy for RT prior to second-line TA; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-EPOCH, rituximab, etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin; R-GV, rituximab, gemcitabine, vinorelbine; R-ICE, rituximab, ifosfamide, carboplatin, etoposide; Rx, treatment; tMDS, treatment-associated myelodysplasia; TOX, toxicity; TTP, time to progression; UNK, unknown; V, venetoclax; VEN, ventoclax.
Outcomes for patients who developed PD on a second-line TA (BTKi or venetoclax). (A) Individual patient characteristics and timelines of outcomes and treatments after the development of PD on either BTKi or venetoclax used sequentially as a second-line TA. Arrows at the end of lanes indicate ongoing survival at last follow-up, circles indicate death, and other treatments have specific symbols as indicated. The columns on the left indicate clinicopathological variables at time of progression on the second-line agent. Gray fill indicates presence of a variable; gray horizontal line indicates new genetic lesion at time of progression on second-line TA; white fill indicates absence; patients who were treated for RT prior to the second-line TA are denoted by a red fill. ^The patient in lane 14 ceased venetoclax in measurable residual disease–positive CR after 6 months, resumed for progressive CLL 12 months later, then subsequently progressed while on drug with a total duration of disease control with venetoclax of 51 months. *Gray fill indicates detection of resistance mutation at any time after exposure to TA; white fill, untested or not detected. (B) OS after the development of PD on a second-line TA. Curves represent outcomes for the overall cohort (green), patients with progressive CLL on a second-line TA (blue, dashed), and patients with RT on second-line TA (red, dashed). (C) OS after the development of PD on a second-line TA, stratified by prior sequencing of TAs. Curves represent the outcomes for patients who receiving BTKi’s then venetoclax (blue) or venetoclax then BTKi’s (red). AutoSCT, autologous stem cell transplant for RT prior to second-line TA; AZA, azacitidine; B, BTKi; IBR, ibrutinib; IFRT, involved-field radiotherapy for RT prior to second-line TA; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-EPOCH, rituximab, etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin; R-GV, rituximab, gemcitabine, vinorelbine; R-ICE, rituximab, ifosfamide, carboplatin, etoposide; Rx, treatment; tMDS, treatment-associated myelodysplasia; TOX, toxicity; TTP, time to progression; UNK, unknown; V, venetoclax; VEN, ventoclax.
Twelve patients received venetoclax 100 to 600 mg daily (majority, ≥400 mg daily; n = 7) as their first-line TA (monotherapy, n = 9; concomitant rituximab, n = 3). Best responses were stable disease, partial remission (PR), and complete remission (CR) in 8%, 58%, and 34% of patients, respectively, and median time to progression (TTP) after venetoclax initiation was 24 months (range, 9-94 months). Three patients developed RT on venetoclax, received salvage chemotherapy followed by consolidative autologous stem cell transplant (SCT; n=2) or involved-field radiotherapy (IFRT; n=1), and later commenced BTKi treatment for progressive CLL (ibrutinib, n = 1; zanubrutinib, n = 2). The remaining 9 patients developed progressive CLL on venetoclax and proceeded directly to BTKi (ibrutinib, n = 8; zanubrutinib, n = 1). The objective response rate to second-line BTKi was 84% (PR, 67%; CR, 17%) with a median TTP of 25 months (1-55 months). At PD on second-line BTKi, 4 patients had RT (relapse of previous RT, n=1; first manifestation of RT, n=3) and 8 had progressive CLL.
Five patients received standard-dose covalent BTKi as their first-line TA (ibrutinib, n = 4; zanubrutinib, n = 1). The objective response rate was 80% (all PRs), and median TTP was 24 months (range, 4-42 months). In all 5 patients, PD on first-line BTKi was with CLL, with no cases of RT, and all patients received 400 mg of venetoclax daily (monotherapy) as their next therapy. Three patients had disease response to second-line venetoclax (all PRs), with a median TTP of 23 months (6-29 months). At progression on second-line venetoclax, 2 patients had RT (with synchronous diagnosis of treatment-associated myelodysplasia, n = 1) and 3 had progressive CLL (supplemental Figure 1B).
Treatments and survival outcomes among the 17 patients with PD on second-line TAs are shown in Figure 1A. The whole-cohort median OS after PD on second-line TAs was 3.6 months (95% confidence interval, 2-11 months). Survival did not differ significantly between patients with RT or progressive CLL on second-line TAs (median OS, 3.3 vs 8 months, respectively; P = .341) (Figure 1B). OS did not differ significantly between patients treated first-line with venetoclax or BTKi (median OS, 2.9 vs 5.3, respectively; P = .756) (Figure 1C). Univariate analyses did not identify any significant association between OS and baseline clinicopathological variables, although these were limited by modest numbers (supplemental Table 1). The majority of patients (13 of 17; 76%) have died, predominantly due to PD (RT, n= 8; CLL, n=3; cardiac arrest, n=1; pneumonia [toxicity], n=1). Four patients with PD on second-line TAs remain alive at last follow-up. Three of these surviving patients with limited follow-up received pirtobrutinib (n = 1, alive <2 months postprogression), ibrutinib retreatment (n = 1, alive <2 months postprogression), and a reduced-dose regimen of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone followed by IFRT consolidation (n = 1, alive 10 months postprogression). One patient received idelalisib-rituximab followed by allogeneic SCT (alloSCT) (n = 1; alive 39 months postprogression), representing the only instance of longer-term survival after developing double class-resistant CLL. Interestingly, 2 patients achieved control of double class-resistant CLL with ibrutinib-venetoclax combination therapy for 10 and 15 months, respectively, including 1 patient in whom BCL2, BTK, and PLCγ2 mutations were detectable (Figure 1A lanes 2 and 4).
These data indicate that patients with CLL sequentially resistant to BTKi and venetoclax therapy have a dismal prognosis and a high frequency of RT. Currently available therapeutic options include PI3K inhibitors and chemoimmunotherapy, but these are challenged by issues with efficacy and tolerability.17 Given the poor prognosis after PD on second-line TA therapy, alloSCT should be strongly considered for fit patients with suitable donors at the time of stable remission on second-line TA, especially if CK or TP53 aberrations are present.22,23 Preliminary clinical data for noncovalent BTKi’s and chimeric antigen receptor T-cell therapy are promising, but formal regulatory approval and longer-term follow-up are required before incorporation into routine care.24,25 The patients in this cohort were heavily pretreated with chemoimmunotherapy, and the generalizability of our findings is uncertain for patients who are chemotherapy-naive and received TAs as frontline therapy or in combination. Nevertheless, these data establish double class-resistant CLL as a high-risk clinical entity for which novel therapeutic approaches are urgently needed.
Acknowledgment
The authors thank all of the patients, family members, and staff who participated in the studies.
Authorship
Contribution: J.F.S., A.W.R., C.S.T., T.E.L., and M.A.A. conceived of the project and designed the study; J.F.S., A.W.R., C.S.T., T.E.L., M.A.A., S.M.H., and B.J.K. were responsible for patient care; D.A.W. contributed flow cytometry and molecular data; V.S.L., T.E.L., and E.R.C. collected the data; T.E.L. analyzed the data; P.B. and E.R.T. analyzed and collated the genomic data; T.E.L. and V.S.L. wrote the first version of the manuscript; and all authors reviewed the data and contributed to critical revision of the manuscript.
Conflict-of-interest disclosure: A.W.R., M.A.A., T.E.L., and V.S.L. are employees of the Walter and Eliza Hall Institute of Medical Research, which receives milestone and royalty payments related to venetoclax. A.W.R., M.A.A., and T.E.L. are recipients of a share in royalty payments paid to the Walter and Eliza Hall Institute of Medical Research. S.M.H. has received honoraria from Gilead and nonfinancial assistance from AbbVie. C.S.T. has received honoraria and research funding from AbbVie and Janssen and honoraria from BeiGene. A.W.R. has received research funding from AbbVie, Genentech, Servier, Janssen, and BeiGene. J.F.S. receives research funding from AbbVie, Genentech, Celgene, and Janssen; and is an advisory board member for, and has received honoraria from, AbbVie, Acerta, Celgene, Genentech, Janssen, Roche, Sunesis, and Takeda. M.A.A. has received honoraria from AbbVie, Janssen, AstraZeneca, Novartis, and CSL Behring. T.E.L. has received honoraria from AbbVie. The remaining authors declare no competing financial interests.
Correspondence: Mary Ann Anderson, Department of Clinical Haematology, The Royal Melbourne Hospital and Peter MacCallum Cancer Centre, Melbourne, VIC, Australia; e-mail: manderson@wehi.edu.au.


