Key Points
The IAP antagonist tolinapant acts as an immunomodulatory molecule in TCL in preclinical models and confirmed in patients.
Tolinapant acts on both the innate and adaptive immune system and can be exploited to remodel the tumor immune microenvironment.

Abstract
Tolinapant (ASTX660) is a potent, nonpeptidomimetic antagonist of cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) and X-linked IAP, which is currently being evaluated in a phase 2 study in T-cell lymphoma (TCL) patients. Tolinapant has demonstrated evidence of single-agent clinical activity in relapsed/refractory peripheral TCL and cutaneous TCL. To investigate the mechanism of action underlying the single-agent activity observed in the clinic, we have used a comprehensive translational approach integrating in vitro and in vivo models of TCL confirmed by data from human tumor biopsies. Here, we show that tolinapant acts as an efficacious immunomodulatory molecule capable of inducing complete tumor regression in a syngeneic model of TCL exclusively in the presence of an intact immune system. These findings were confirmed in samples from our ongoing clinical study showing that tolinapant treatment can induce changes in gene expression and cytokine profile consistent with immune modulation. Mechanistically, we show that tolinapant can activate both the adaptive and the innate arms of the immune system through the induction of immunogenic forms of cell death. In summary, we describe a novel role for IAP antagonists as immunomodulatory molecules capable of promoting a robust antitumor immune response in TCL.
Introduction
T-cell lymphomas (TCLs) are a group of lymphoid malignancies originating from mature postthymic T cells and characterized by high clinicopathological diversity and an aggressive course.1 The majority of TCL patients will not achieve prolonged complete remissions with current first-line standard-of-care chemotherapy (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone),2 and very limited treatment options are available for the relapsed/refractory group.3 Thus, there is an unmet clinical need to identify novel therapeutic options to improve the limited prognosis faced by these patients.4
The cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1 and cIAP2), and X-linked IAP (XIAP), are key regulators of apoptosis and survival signaling pathways: XIAP directly inhibits caspases, whereas cIAP1/2 suppress the formation of pro-apoptotic signaling complexes and allow pro-survival signaling.5,6 This leads to suppression of apoptosis through both the extrinsic and intrinsic apoptosis pathways. Because of their role in evasion of apoptosis, IAPs are considered attractive targets for anticancer therapy and several small-molecule antagonists are currently in clinical development with a focus on cancer treatment.7,8 In addition to their antiapoptotic role, IAP proteins have been reported to have a role in immunoregulation through modulation of both innate and adaptive immunity.9 By acting on two of the key hallmarks of cancer, cell survival and immune evasion, IAP antagonists represent a promising therapeutic approach to modulate cell death mechanisms, while simultaneously unleashing the power of the immune system to eradicate the tumor.10
Tolinapant (ASTX660) is a novel nonpeptidomimetic, small-molecule antagonist of cIAP1/2 and XIAP discovered using fragment-based drug design.11,12 Tolinapant is currently being evaluated in a phase 1-2 study in patients with advanced solid tumors or lymphoma (NCT02503423). In the ongoing phase 2 ASTX660-01 study, tolinapant has demonstrated preliminary evidence of single-agent clinical activity in relapsed/refractory peripheral T-cell lymphoma (PTCL) and cutaneous T-cell lymphoma patients.13,14
In this study, we aim to further our understanding of tolinapant’s mechanism of action in TCL using a combination of preclinical models and patient-derived biopsies. Together with its well-characterized pro-apoptotic effect, we describe a novel role for tolinapant as an immunomodulatory molecule capable of promoting an antitumor immune response in TCL.
Materials and methods
Animals
AKR/J, CB17 SCID, and BALB/c mice were used to generate human xenograft and syngeneic TCL models. The care and treatment of animals were in accordance with the United Kingdom Coordinating Committee for Cancer Research guidelines and with the United Kingdom Animals (Scientific Procedures) Act 1986.15,16
Primary human cells
Coculture experiments were performed using peripheral blood mononuclear cells (PBMCs) isolated from healthy human donors. All human donors provided written consent and materials were used in accordance with Human Tissue Authority.
Clinical study samples
Tumor biopsies and plasma from subjects enrolled in the phase 1-2 Study of the Safety, Pharmacokinetics, and Preliminary Activity of ASTX660 in Subjects with Advanced Solid Tumors and Lymphomas (NCT02503423) were collected at screening and on treatment. All subjects provided written informed consent for their samples to be stored and used for research purposes. The study protocol was approved by institutional review boards or independent ethics committees prior to initiating the study.
Detailed information about cell lines, antibodies, and chemicals, as well as in vitro and in vivo methods are described in supplemental Materials and Methods.
Results
Tolinapant target engagement and induction of cell death in in vitro TCL models
We previously reported that treatment with tolinapant led to antagonism of XIAP and cIAP1 and tumor necrosis factor-α (TNF-α)-dependent induction of apoptosis in various cancer cell lines in vitro.12 Here, we extended these observations in TCL-relevant in vitro models. Tolinapant treatment both antagonized SMAC binding to XIAP and led to cIAP1 degradation in a dose-dependent fashion in mouse BW5147.G.1.4 (BW5147) and human HH TCL cells (Figure 1A-B) as well as in 2 additional mouse TCL cell lines (supplemental Figure 1A). cIAP2 antagonism was also demonstrated in the human HH cell line (Figure 1B). The effects of tolinapant treatment on the viability of mouse and human TCL cell lines in the absence or presence of exogenous TNF-α were investigated. The cell lines tested ranged in response to tolinapant (Figure 1C-F). All 4 mouse cell lines were sensitive to tolinapant in the presence of TNF-α; whereas human cell lines, which had low basal expression of TNFR1 (supplemental Figure 1B), remained mostly insensitive. The mouse cell line, BW5147, was identified as the most sensitive in the absence of exogenous TNF-α (Figure 1C). This response was not ablated by neutralization of TNF-α (supplemental Figure 1C). Western blotting experiments using BW5147 cells treated with tolinapant demonstrated an increase in apoptotic markers as well as increased markers of DNA damage, which was augmented by inclusion of TNF-α (Figure 1G). Increased cell death was confirmed in BW5147 cells treated for 48 hours with tolinapant by cytometry (Figure 1H; supplemental Figure 1D).
![Tolinapant target engagement and induction of cell death in in vitro TCL models. (A) cIAP1 (by level reduction) and XIAP (by XIAP:SMAC complex immunoprecipitation) antagonism in mouse BW5147 cells treated with tolinapant for 2 hours. (B) cIAP1/2 and XIAP antagonism in human HH cells treated with tolinapant for 2 hours. (C) Cell viability (CellTiter-Glo [CTG]) assay data after 72 hours’ treatment of 4 mouse TCL cell lines with tolinapant alone. (D) As for panel C, except treated with tolinapant plus 10 ng/mL mouse TNF-α. (E) Cell viability (CTG) assay data after 72 hours of treatment of 5 human TCL cell lines with tolinapant alone. (F) As for panel E, except treated with tolinapant plus 10 ng/mL human TNF-α. (G) Western blots of BW5147 cell lysates after 24-hour treatment with tolinapant or tolinapant plus 10 ng/mL TNF-α. (H) BW5147 viability after 24-hour treatment with tolinapant or tolinapant plus TNF-α by cytometry (propidium iodide [PI] and cleaved caspase-3 dual staining [CC3]).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/20/10.1182_bloodadvances.2020003955/7/m_advancesadv2020003955f1.png?Expires=1768262439&Signature=FdX2yt8lpWthI0aA86h4K3alBXX4VB9t3q1JDKWUAx0UTmO~8SyPnvi7DZXd1IvhE9xZT4z5nOXNM3xmY6gxls1~KH5diUG8GfW-OXG32uB3ZsOD~ODV4ltteKqDYs8r48nvmJGBktICH~vyb9uKKikreSr9X~x2PpiXa~r0q30zrSBxIunzJyUdk8I2hdZkAps9zPK7sYyVRA4QAduReLNU6Vmdz7f5v4xr5C8tJOAuJNRgRfclWzbvXyIYVjUzy6nM8eWFZ-Xpo6vD7AfIAVm4GH2c2p1dpixkUUGmWSat8x7mjnEbxypLIOcPkT3VFX5RUph6zYvDrc0YpByhXw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Tolinapant target engagement and induction of cell death in in vitro TCL models. (A) cIAP1 (by level reduction) and XIAP (by XIAP:SMAC complex immunoprecipitation) antagonism in mouse BW5147 cells treated with tolinapant for 2 hours. (B) cIAP1/2 and XIAP antagonism in human HH cells treated with tolinapant for 2 hours. (C) Cell viability (CellTiter-Glo [CTG]) assay data after 72 hours’ treatment of 4 mouse TCL cell lines with tolinapant alone. (D) As for panel C, except treated with tolinapant plus 10 ng/mL mouse TNF-α. (E) Cell viability (CTG) assay data after 72 hours of treatment of 5 human TCL cell lines with tolinapant alone. (F) As for panel E, except treated with tolinapant plus 10 ng/mL human TNF-α. (G) Western blots of BW5147 cell lysates after 24-hour treatment with tolinapant or tolinapant plus 10 ng/mL TNF-α. (H) BW5147 viability after 24-hour treatment with tolinapant or tolinapant plus TNF-α by cytometry (propidium iodide [PI] and cleaved caspase-3 dual staining [CC3]).
Tolinapant target engagement and induction of cell death in in vitro TCL models. (A) cIAP1 (by level reduction) and XIAP (by XIAP:SMAC complex immunoprecipitation) antagonism in mouse BW5147 cells treated with tolinapant for 2 hours. (B) cIAP1/2 and XIAP antagonism in human HH cells treated with tolinapant for 2 hours. (C) Cell viability (CellTiter-Glo [CTG]) assay data after 72 hours’ treatment of 4 mouse TCL cell lines with tolinapant alone. (D) As for panel C, except treated with tolinapant plus 10 ng/mL mouse TNF-α. (E) Cell viability (CTG) assay data after 72 hours of treatment of 5 human TCL cell lines with tolinapant alone. (F) As for panel E, except treated with tolinapant plus 10 ng/mL human TNF-α. (G) Western blots of BW5147 cell lysates after 24-hour treatment with tolinapant or tolinapant plus 10 ng/mL TNF-α. (H) BW5147 viability after 24-hour treatment with tolinapant or tolinapant plus TNF-α by cytometry (propidium iodide [PI] and cleaved caspase-3 dual staining [CC3]).
Tolinapant induces durable regressions in an in vivo syngeneic model of TCL
To evaluate the effects of tolinapant treatment in vivo, we used 2 human xenograft models. Daily oral treatment with tolinapant at a well-tolerated dose (supplemental Figure 2A-B) on the HH (Figure 2A) and SUP-T1 (Figure 2B) models significantly slowed tumor growth (supplemental Figure 2C-D). Western blot analysis from tumors extracted from mice that received a single oral dose of tolinapant showed degradation of cIAP1 and cIAP2, as expected (Figure 2C). Upregulation of apoptosis markers was also detectable as early as 2 hours postdose, confirming the ability of tolinapant to induce cell death in vivo.
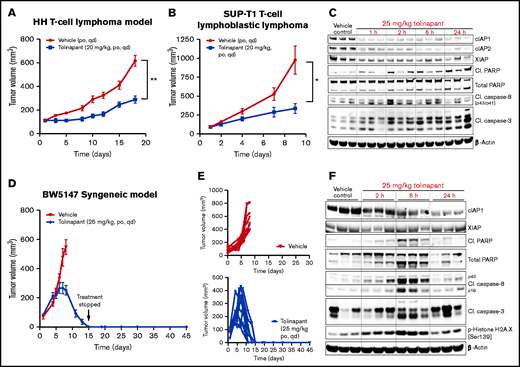
Tolinapant induces durable regressions in an in vivo syngeneic model of TCL. (A) Mice bearing ∼100 mm3 HH tumors were treated with vehicle or with 20 mg/kg tolinapant (daily oral for 18 days). Error bars, mean ± standard error of the mean (SEM). n = 10 per group. Two-way analysis of variance (ANOVA): **P < .01. (B) Mice bearing ∼100 mm3 SUP-T1 tumors were treated with vehicle or with 20 mg/kg tolinapant (daily oral for 9 days). Error bars, mean ± SEM. n = 8 per group. Two-way ANOVA: *P < .05. (C) Mice bearing HH xenograft tumors received a single oral dose of tolinapant at 25 mg/kg. Animals were sacrificed at the indicated time points, and protein levels in tumors were measured by immunoblotting of whole-tumor lysates. Each sample represents individual tumors. (D) Mice bearing ∼50 mm3 BW5147 tumors were treated with vehicle or with 25 mg/kg tolinapant (daily oral). Error bars, mean ± SEM. n = 10 per group. (E) Individual BW5147 tumor volume plots from mice treated with vehicle (top) or 25 mg/kg tolinapant (bottom) (n = 10 per group). (F) Mice bearing BW5147 tumors received a single oral dose of tolinapant at 25 mg/kg. Animals were euthanized at the indicated time points, and protein levels in tumors were measured by immunoblotting of whole-tumor lysates. Each sample represents individual tumors.
Tolinapant induces durable regressions in an in vivo syngeneic model of TCL. (A) Mice bearing ∼100 mm3 HH tumors were treated with vehicle or with 20 mg/kg tolinapant (daily oral for 18 days). Error bars, mean ± standard error of the mean (SEM). n = 10 per group. Two-way analysis of variance (ANOVA): **P < .01. (B) Mice bearing ∼100 mm3 SUP-T1 tumors were treated with vehicle or with 20 mg/kg tolinapant (daily oral for 9 days). Error bars, mean ± SEM. n = 8 per group. Two-way ANOVA: *P < .05. (C) Mice bearing HH xenograft tumors received a single oral dose of tolinapant at 25 mg/kg. Animals were sacrificed at the indicated time points, and protein levels in tumors were measured by immunoblotting of whole-tumor lysates. Each sample represents individual tumors. (D) Mice bearing ∼50 mm3 BW5147 tumors were treated with vehicle or with 25 mg/kg tolinapant (daily oral). Error bars, mean ± SEM. n = 10 per group. (E) Individual BW5147 tumor volume plots from mice treated with vehicle (top) or 25 mg/kg tolinapant (bottom) (n = 10 per group). (F) Mice bearing BW5147 tumors received a single oral dose of tolinapant at 25 mg/kg. Animals were euthanized at the indicated time points, and protein levels in tumors were measured by immunoblotting of whole-tumor lysates. Each sample represents individual tumors.
Several reports have linked IAP antagonists to an immunomodulatory anticancer effect.9,17–21 To investigate a possible novel immunomodulatory role of tolinapant in TCL, we set out to test the effects of tolinapant treatment in the context of a fully functional immune system using in vivo syngeneic models of TCL. Based on our in vitro results, the tolinapant single-agent sensitive BW5147 cell line was selected to establish an in vivo model. Mice bearing subcutaneous BW5147 tumors were treated orally with tolinapant at a well-tolerated dose (supplemental Figure 2E). BW5147 tumors continued to grow for the first 7 days of treatment and then started to regress. By day 15, none of the tumors were palpable (10/10 mice showing complete regressions) (Figure 2D). Treatment was stopped after day 15, and all mice remained tumor-free 30 days later (Figure 2E). Single-dose pharmacodynamic experiments confirmed that systemic administration of tolinapant leads to degradation of its primary target protein, cIAP1, and upregulation of cell death markers (Figure 2F). The antitumor activity observed in the BW5147 syngeneic model suggested that immune-mediated tumor killing could play a role in the in vivo antitumor effects seen during tolinapant treatment.
Tolinapant shows costimulatory activity in human effector T cells and promotes immune-mediated tumor killing
To determine if tolinapant treatment could stimulate effector T-cell function, we used superantigen staphylococcal enterotoxin B (SEB) stimulation as a model of T-cell activation. PBMCs from healthy donors stimulated with SEB in the presence of tolinapant produced increased levels of interleukin-2 (IL-2), in a dose-dependent manner (Figure 3A-B), achieving a level similar to that induced by an anti-PD1 antibody (supplemental Figure 3A-B).
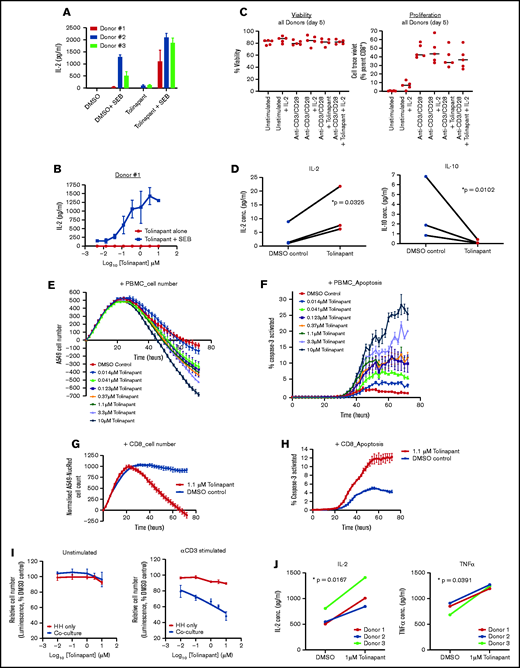
Tolinapant promotes immune-mediated tumor killing. (A) Secreted IL-2 level measured by Luminex assay after 5-day treatment of PBMC (3 donors) with 1 µM tolinapant in the presence or absence of 1 µg/mL SEB. Paired statistical analysis of the “dimethyl sulfoxide (DMSO) + SEB” vs “tolinapant + SEB” group, P = .0005 (paired t test). (B) As for panel A, except full-dose response to tolinapant in the presence or absence of SEB. (C) Unstimulated and stimulated CD8+ T-cell (from 5 donors) viability (viability dye eFluor 780) or proliferation (CellTrace Violet) after 5-day treatment with or without 10 µM tolinapant. (D) IL-2 or IL-10 level measured by Luminex assay after 5-day treatment of anti-CD3/CD28 stimulated CD8+ T cells (from 3 donors) with 1 µM tolinapant. *P < .05 (ratio paired t test). (E) A549-NucLight Red cell killing by anti CD3-activated PBMCs in the presence of tolinapant by IncuCyte. (F) Measurement of activated caspase-3 in A549-NucLight Red cells in F by IncuCyte. (G) A549-NucLight Red cell killing by anti-CD3/CD28-activated CD8+ T cells in the presence of tolinapant by IncuCyte. (H) Measurement of activated caspase-3 in A549-NucLight Red cells in H by IncuCyte. (I) Percentage cell viability of HH cells alone or HH cells cocultured with PBMCs treated with increasing concentrations of tolinapant without (left) or with (right) anti-CD3 stimulation after 48 hours (n = 3). (J) Secreted IL-2 and TNF-α levels measured by MSD assay from anti-CD3-stimulated HH-PBMC coculture assays treated with DMSO or 1 µM tolinapant for 72 hours. *P < .05 (ratio paired t test).
Tolinapant promotes immune-mediated tumor killing. (A) Secreted IL-2 level measured by Luminex assay after 5-day treatment of PBMC (3 donors) with 1 µM tolinapant in the presence or absence of 1 µg/mL SEB. Paired statistical analysis of the “dimethyl sulfoxide (DMSO) + SEB” vs “tolinapant + SEB” group, P = .0005 (paired t test). (B) As for panel A, except full-dose response to tolinapant in the presence or absence of SEB. (C) Unstimulated and stimulated CD8+ T-cell (from 5 donors) viability (viability dye eFluor 780) or proliferation (CellTrace Violet) after 5-day treatment with or without 10 µM tolinapant. (D) IL-2 or IL-10 level measured by Luminex assay after 5-day treatment of anti-CD3/CD28 stimulated CD8+ T cells (from 3 donors) with 1 µM tolinapant. *P < .05 (ratio paired t test). (E) A549-NucLight Red cell killing by anti CD3-activated PBMCs in the presence of tolinapant by IncuCyte. (F) Measurement of activated caspase-3 in A549-NucLight Red cells in F by IncuCyte. (G) A549-NucLight Red cell killing by anti-CD3/CD28-activated CD8+ T cells in the presence of tolinapant by IncuCyte. (H) Measurement of activated caspase-3 in A549-NucLight Red cells in H by IncuCyte. (I) Percentage cell viability of HH cells alone or HH cells cocultured with PBMCs treated with increasing concentrations of tolinapant without (left) or with (right) anti-CD3 stimulation after 48 hours (n = 3). (J) Secreted IL-2 and TNF-α levels measured by MSD assay from anti-CD3-stimulated HH-PBMC coculture assays treated with DMSO or 1 µM tolinapant for 72 hours. *P < .05 (ratio paired t test).
Because CD8+ T cells play a critical role in antitumor immunity and their function has been reported to be affected by IAP antagonism,17,19,21,22 we next explored the effects of tolinapant treatment on stimulated CD8+ T cells freshly isolated from healthy donors. Although viability and proliferation of CD8+ T cells were unaffected during these studies (Figure 3C), tolinapant treatment increases secretion of IL-2 and decreases expression of IL-10 (Figure 3D). Collectively these findings suggest that tolinapant treatment can augment normal lymphocyte costimulation.
We then investigated whether tolinapant-induced activation of T cells also leads to increased functional cytotoxic activity with in vitro coculture killing assays using a tolinapant-resistant cell line (supplemental Figure 3C-D). Activated PBMCs from healthy donors were added to a monolayer of A549 cancer cells and incubated with tolinapant. Treatment led to dose-dependent decrease of A549 tumor cells overtime (Figure 3E) and to a corresponding increase of cancer cell apoptosis (Figure 3F). In contrast, tolinapant had no effects on A549 cells alone or on A549 in the presence of unstimulated PBMC, confirming that the observed effect is due to immune-mediated killing (supplemental Figure 3E). These studies were repeated with isolated CD8 T cells and natural killer cells to better understand the cytotoxic mechanism involved. In the presence of tolinapant, both activated CD8 (Figure 3G-H) and natural killer cells (supplemental Figure 3F-G) showed increased killing capability leading to a strong decrease of A549 tumor cells overtime and to a corresponding increase of cancer cell apoptosis. Blocking antibodies to TNF-α or interferon-γ (IFN-γ) partially inhibit tolinapant-driven immune killing with further inhibition achieved by combination of TNF-α and IFN-γ blockade (supplemental Figure 3H), suggesting that tolinapant-driven tumor cell death requires both TNF-α and IFN-γ, most likely produced by activated PBMCs.
To corroborate our results in a more disease-relevant system, we established a human TCL killing assay using the HH cell line. Treatment of HH cells with tolinapant in the presence of activated PBMCs from healthy donors led to a significant decrease in tumor cell viability in a dose-dependent fashion (Figure 3I). Tolinapant had no effect on HH cancer cells alone or in coculture with unstimulated PBMCs, confirming that the observed effect is due to immune-mediated killing. Moreover, analysis of the coculture supernatant showed that tolinapant treatment led to an increase of IL-2 and TNF-α secretion, 2 cytokines that have been shown to promote cytotoxic T-cell response (Figure 3J). Blocking antibodies to TNF-α and/or IFN-γ partially inhibit tolinapant-driven immune killing (supplemental Figure 3I), suggesting that tolinapant-driven tumor cell death is, at least in part, occurring via increased production of TNF-α and IFN-γ by activated PBMCs. Further, in HH cell coculture assays using the IL-15–stimulated human natural killer cell line NK92, we observed similar tolinapant dose-dependent killing of the HH tumor target cells (supplemental Figure 3J).
Overall, these findings suggest that tolinapant can broadly costimulate lymphoid immune cell populations involved in antitumor responses, leading to specific changes in cytokine profiles and increased cytotoxicity.
Tolinapant requires an intact immune system to generate long-term tumor regressions
To provide an initial insight into the immunomodulatory action of tolinapant in vivo, we analyzed the effects of tolinapant treatment on gene expression in BW5147 murine tumors. Tumor RNA was purified after 9 days of treatment and transcriptional profiling of a panel of immune-associated genes was obtained. An increased cytotoxic cell signature was detected in tolinapant-treated tumors (Figure 4A; supplemental Figure 4A-C). In addition, tolinapant significantly increased intratumoral expression levels of Gzmb, Gzme, and Prf-1 (Figure 4B; supplemental Figure 4D), key marker genes expressed by cytotoxic lymphocytes and required for their killing potential. We next performed immunofluorescence analysis to validate these results. Consistent with RNA profiling, a significant increase in CD8+ cells was detectable in tumors from tolinapant-treated mice, compared with control (Figure 4C). Together with increased cytotoxic cell activity, tolinapant also promoted the establishment of a pro-inflammatory tumor microenvironment characterized by increased interferon signaling, immune infiltration, and costimulatory signatures. In contrast, genes linked to cell proliferation were downregulated (Figure 4D; supplemental Figure 4E). Collectively, these results suggest that a robust effector T-cell response is elicited by tolinapant treatment in vivo in the BW5147 syngeneic TCL model.
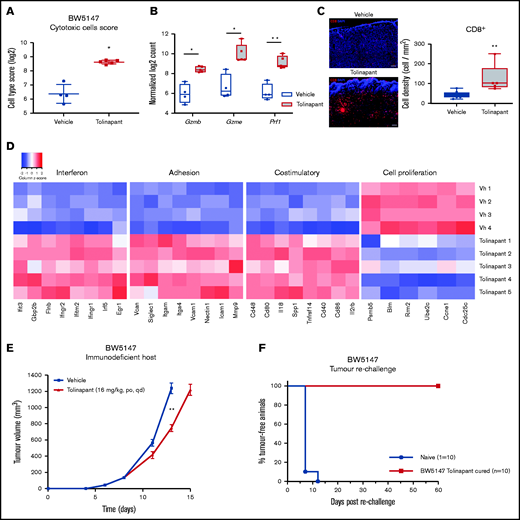
Tolinapant requires an intact immune system to generate long-term tumor regressions. (A) Cytotoxic cells score in BW5147 subcutaneous tumors in vehicle and tolinapant-treated mice (log2 scale). *P < .05 (2-sided Welch t test adjusted using the Benjamini and Yekutieli method). (B) Intratumoral expression of Gzmb, Gzme, and Prf1. *P < .05, **P < .01 (adjusted P value). (C) Representative immunofluorescent images and boxplot showing quantification of CD8+ cells in tumor sections. **P < .001 (Mann-Whitney test). Tumor formalin-fixed paraffin-embedded sections were costained with fluorochrome-conjugated anti-CD8 (red) antibody and DAPI (blue). (D) Heatmap of the selected genes that are differentially expressed (adjusted P value < .05) between vehicle and tolinapant-treated BW5147 tumors, displayed as gene-wise z scores (log2 normalized expression). Selected genes were obtained from gene expression signatures associated with different pathways scored with NanoString Advanced Analysis GSA module. (E) Immunodeficient CB17 SCID mice were treated with vehicle or with 16 mg/kg tolinapant (daily oral) 4 days after cell injection (day 0). Error bars, mean ± SEM. n = 10 per group. Two-way ANOVA: **P < .01. (F) Kaplan-Meier curve depicting percentage of tumor-free animals after rechallenge of naïve or tolinapant-cured mice with BW5147 cells. n = 10 per group.
Tolinapant requires an intact immune system to generate long-term tumor regressions. (A) Cytotoxic cells score in BW5147 subcutaneous tumors in vehicle and tolinapant-treated mice (log2 scale). *P < .05 (2-sided Welch t test adjusted using the Benjamini and Yekutieli method). (B) Intratumoral expression of Gzmb, Gzme, and Prf1. *P < .05, **P < .01 (adjusted P value). (C) Representative immunofluorescent images and boxplot showing quantification of CD8+ cells in tumor sections. **P < .001 (Mann-Whitney test). Tumor formalin-fixed paraffin-embedded sections were costained with fluorochrome-conjugated anti-CD8 (red) antibody and DAPI (blue). (D) Heatmap of the selected genes that are differentially expressed (adjusted P value < .05) between vehicle and tolinapant-treated BW5147 tumors, displayed as gene-wise z scores (log2 normalized expression). Selected genes were obtained from gene expression signatures associated with different pathways scored with NanoString Advanced Analysis GSA module. (E) Immunodeficient CB17 SCID mice were treated with vehicle or with 16 mg/kg tolinapant (daily oral) 4 days after cell injection (day 0). Error bars, mean ± SEM. n = 10 per group. Two-way ANOVA: **P < .01. (F) Kaplan-Meier curve depicting percentage of tumor-free animals after rechallenge of naïve or tolinapant-cured mice with BW5147 cells. n = 10 per group.
To further confirm tolinapant’s contribution in enhancing an antitumor immune response, BW5147 tumors were grown in immunodeficient mice lacking functional B and T cells. BW5147 tumors grown in an immunocompromised host and treated with doses of tolinapant chosen to achieve equivalent exposures in the 2 mouse strains, only showed a modest tumor growth inhibition after treatment (Figure 4E), suggesting that an intact immune system is required to drive the tumor regressions seen in the immune competent mice. To further investigate the contribution of the adaptive immune system in tolinapant-mediated response, a tumor-rechallenge study was conducted in mice whose tumors had regressed completely following tolinapant treatment. These mice were completely resistant to tumor engraftment (Figure 4F), pointing to an antitumor memory effect mediated by the adaptive immune system. Collectively, these results demonstrate that tolinapant is acting as an immunomodulatory agent in vivo and is able to generate stable long-term tumor regressions only in the presence of an intact immune system.
Tolinapant promotes activation of the innate immune system by inducing necroptosis and immunogenic cell death in tumor cells
The mechanisms by which tolinapant induces an enhanced adaptive response were further explored by gene expression analysis in 2 in vivo tumor models, BW5147 and HH. An increase of a myeloid signature upon tolinapant dosing was observed in both models (Figure 5A). Further pathway analysis identified a strong upregulation of several genes linked to myeloid lineage and antigen presentation together with several chemokines and cytokines (Figure 5B; supplemental Figure 5A-B). Taken together, these data suggest that tolinapant treatment affects the intratumoral innate immune infiltrate in vivo. Because IAP antagonists have been shown to affect the macrophage lineage,23 we examined the impact of tolinapant on macrophages in greater detail using primary human monocytes in vitro cultures. An increase in phenotypic markers linked to myeloid activation was detected in primary human CD14+ M0 monocytes cultured for 6 days in the presence of 1 µM tolinapant (Figure 5C). The impact of tolinapant on macrophage polarization was then evaluated. An increase in M1 phenotypic markers (Figure 5D) over control was observed after tolinapant treatment in both M1 and M2 macrophage populations, suggesting that tolinapant can promote M1-like macrophage polarization in vitro.
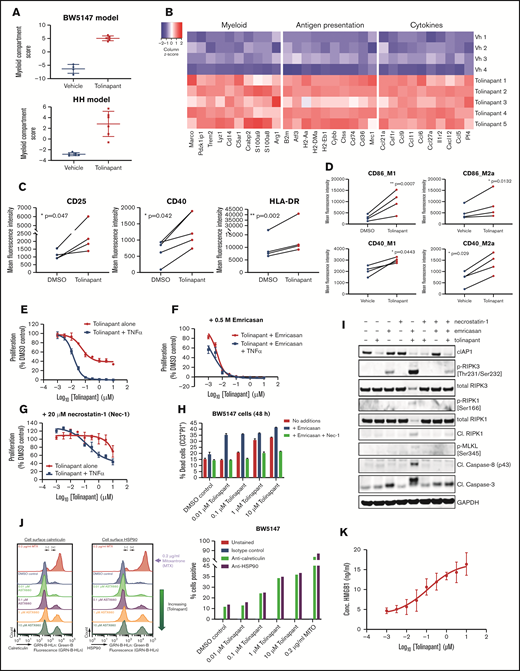
Tolinapant promotes activation of the innate immune system by inducing necroptosis and immunogenic cell death in tumor cells. (A) Myeloid pathway score for the BW5147 model (top) and the HH model (bottom) calculated using NanoString Advanced Analysis pathway scores module. (B) Heatmap of the expression of selected genes that are differentially expressed between vehicle and tolinapant-treated BW5147 tumors (adjusted P < .05), displayed as gene-wise z-scores (log2 normalized expression). Selected genes were obtained from gene expression signatures associated with different pathways scored with NanoString Advanced Analysis GSA module. (C) Expression of CD25, CD40, and HLA-DR on M0 macrophages at day 6 of treatment with vehicle (DMSO) or 1 µM tolinapant analyzed by flow cytometry (3 donors). *P < .05, **P < .01 (ratio paired t test). (D) Expression of CD86 (top) and CD40 (bottom) on polarized M1 and M2 macrophages at day 8 of treatment with vehicle (DMSO) or 1 µM tolinapant analyzed by flow cytometry (3 donors). *P < .05, **P < .001 (ratio paired t test). (E) Effect of tolinapant or tolinapant + 10 ng/mL mouse TNF-α on BW5147 cell viability by CTG assay after 72 hours. (F) As for panel E, except addition of 0.5 µM emricasan to inhibit caspase-8. (G) As for panel E, except addition of 20 µM Necrostatin-1 (Nec-1) to inhibit RIPK1. (H) Viability of BW5147 cells by cytometry (PI and CC3) after 48 hours’ treatment with tolinapant alone or with 0.5 µM emricasan or with 0. 5 µM emricasan plus 20 µM Nec-1. (I) Western blots of BW5147.1.4 cell lysates prepared after 48-hour treatment with tolinapant alone or with 0.5 µM emricasan or with 0. 5 µM emricasan plus 20 µM Nec-1. (J) BW5147 cell surface calreticulin or HSP90 staining analyzed by cytometry after 24-hour treatment with tolinapant or 0.2 µg/mL mitoxantrone. Data shown as flow cytometry histograms gated on total cells. (K) HMGB1 measured by enzyme-linked immunosorbent assay in BW5147 cell supernatants after 24-hour treatment with tolinapant.
Tolinapant promotes activation of the innate immune system by inducing necroptosis and immunogenic cell death in tumor cells. (A) Myeloid pathway score for the BW5147 model (top) and the HH model (bottom) calculated using NanoString Advanced Analysis pathway scores module. (B) Heatmap of the expression of selected genes that are differentially expressed between vehicle and tolinapant-treated BW5147 tumors (adjusted P < .05), displayed as gene-wise z-scores (log2 normalized expression). Selected genes were obtained from gene expression signatures associated with different pathways scored with NanoString Advanced Analysis GSA module. (C) Expression of CD25, CD40, and HLA-DR on M0 macrophages at day 6 of treatment with vehicle (DMSO) or 1 µM tolinapant analyzed by flow cytometry (3 donors). *P < .05, **P < .01 (ratio paired t test). (D) Expression of CD86 (top) and CD40 (bottom) on polarized M1 and M2 macrophages at day 8 of treatment with vehicle (DMSO) or 1 µM tolinapant analyzed by flow cytometry (3 donors). *P < .05, **P < .001 (ratio paired t test). (E) Effect of tolinapant or tolinapant + 10 ng/mL mouse TNF-α on BW5147 cell viability by CTG assay after 72 hours. (F) As for panel E, except addition of 0.5 µM emricasan to inhibit caspase-8. (G) As for panel E, except addition of 20 µM Necrostatin-1 (Nec-1) to inhibit RIPK1. (H) Viability of BW5147 cells by cytometry (PI and CC3) after 48 hours’ treatment with tolinapant alone or with 0.5 µM emricasan or with 0. 5 µM emricasan plus 20 µM Nec-1. (I) Western blots of BW5147.1.4 cell lysates prepared after 48-hour treatment with tolinapant alone or with 0.5 µM emricasan or with 0. 5 µM emricasan plus 20 µM Nec-1. (J) BW5147 cell surface calreticulin or HSP90 staining analyzed by cytometry after 24-hour treatment with tolinapant or 0.2 µg/mL mitoxantrone. Data shown as flow cytometry histograms gated on total cells. (K) HMGB1 measured by enzyme-linked immunosorbent assay in BW5147 cell supernatants after 24-hour treatment with tolinapant.
IAP antagonists have been shown to promote various forms of immunogenic cell death,24,25 which could lead to increased activation of the innate immune system.26,27 To understand whether the mechanism of BW5147 cell death induced by tolinapant was playing a role in the enhanced myeloid activation seen in vitro and in vivo, we investigated whether BW5147 cells undergo necroptosis in vitro. Emricasan, a caspase 8 inhibitor, enhanced tolinapant-induced cell death (Figure 5E-F,H), suggesting a nonapoptotic mechanism. On the other hand, inclusion of the RIPK1 inhibitor necrostatin-1 (Nec-1), led to increased viability (Figure 5E,G-H), suggesting a possible role for necroptosis in mediating the observed cell death. Western blot analysis confirmed that necroptosis could be initiated by tolinapant in the presence of emricasan as shown by increased expression of the necroptosis markers phospho-RIPK3 and phospho-MLKL, which was blocked by Nec-1 (Figure 5I). High basal level expression of RIPK3 was measured in all 4 mouse TCL cell lines (supplemental Figure 5C). Real-time microscopy demonstrated that cell permeabilization was augmented by incubation of BW5147 cells with tolinapant in the presence of emricasan, further pointing to an involvement of necroptosis (supplemental Figure 5D). Activation of immunogenic cell death pathways leads to activation of the innate immune system via release of damage-associated molecular patterns (DAMPs). BW5147 cells released several DAMPs after exposure to tolinapant in vitro, as shown by increased cell surface levels of calreticulin and HSP90 expression (Figure 5J; supplemental Figure 5E) and by HMGB1 release into the cell supernatant (Figure 5K; supplemental Figure 5F). Together, our results indicate that tolinapant induces DAMPs release via induction of immunogenic forms of cell death in vitro.
Tolinapant promotes antigen-specific responses via induction of necroptosis in an in vivo model of T-cell lymphoma
To investigate whether the promotion of immunogenic cell death/necroptosis seen in vitro was also evident in vivo, the short-term effects of tolinapant treatment on BW5147 tumors were analyzed. Western blot analysis of tumors from mice that received a single oral dose of tolinapant showed a strong increase in necroptosis markers (Figure 6A). Additionally, increased protein expression of immunogenic cell death markers and DAMPs were detected (Figure 6B). HMGB1 serum levels have been used as a biomarker for necroptosis in vivo.28,29 Importantly, after 5 days of tolinapant treatment, HMGB1 levels in the plasma of mice were significantly higher compared with vehicle-treated control animals (Figure 6C). These results indicate that tolinapant is capable of inducing necroptosis and DAMPs release in vivo.

Tolinapant promotes antigen-specific responses via induction of necroptosis in vivo. (A) Western blots of BW5147 syngeneic tumor lysates prepared 2, 6, or 24 hours after a single dose of 25 mg/kg tolinapant (compared with vehicle control) to determine necroptosis biomarker pharmacodynamics. (B) As for panel A, except western blots of immunogenic cell death biomarkers. (C) HMGB1 measured by enzyme-linked immunosorbent assay in plasma of BW5147 tumor-bearing animals after 5 days of treatment with 25 mg/kg tolinapant. *P < .05 (Unpaired t test with Welch’s correction). (D) Kaplan-Meier curve showing the evolution of tumor incidence (as percentage of tumor-free animals) over time in the BW5147 vaccination experiment. Cells killed with mitoxantrone (MITO) were used as a positive control. Mice injected with phosphate-buffered saline only (PBS) or cells killed by tolinapant + TNF-α (tolinapant + TNF-α) cohorts, n = 10 per group. MITO, n = 8 per group.
Tolinapant promotes antigen-specific responses via induction of necroptosis in vivo. (A) Western blots of BW5147 syngeneic tumor lysates prepared 2, 6, or 24 hours after a single dose of 25 mg/kg tolinapant (compared with vehicle control) to determine necroptosis biomarker pharmacodynamics. (B) As for panel A, except western blots of immunogenic cell death biomarkers. (C) HMGB1 measured by enzyme-linked immunosorbent assay in plasma of BW5147 tumor-bearing animals after 5 days of treatment with 25 mg/kg tolinapant. *P < .05 (Unpaired t test with Welch’s correction). (D) Kaplan-Meier curve showing the evolution of tumor incidence (as percentage of tumor-free animals) over time in the BW5147 vaccination experiment. Cells killed with mitoxantrone (MITO) were used as a positive control. Mice injected with phosphate-buffered saline only (PBS) or cells killed by tolinapant + TNF-α (tolinapant + TNF-α) cohorts, n = 10 per group. MITO, n = 8 per group.
An in vivo vaccination assay was used to confirm the role of tolinapant as a novel immunogenic cell death (ICD) inducer.30,31 Subcutaneous implantation of BW5147 cells killed in vitro by tolinapant in the presence of TNF-α completely protected immunocompetent AKR/J mice against rechallenge with live BW5147 tumor cells when injected 1 week later into the opposite flank (Figure 6D). This effect was comparable to a positive control, in which BW5147 cells killed by mitoxantrone, a known inducer of ICD,32,33 were used. The in vivo vaccination experiment demonstrated that tolinapant can act as a bona fide ICD inducer, which is able to activate a specific and long-term antitumor immune response via release of DAMPs.
Tolinapant treatment has an immunomodulatory effect in TCL patients
To determine if our preclinical findings are relevant in the clinical setting, we analyzed biopsies from 6 PTCL subjects available from the ongoing clinical trial. For each of the 6 patients, biopsies were taken before and during tolinapant treatment and transcriptomic profiles were determined. Cell profiling analysis was used to investigate the effect of tolinapant treatment on immune cell populations (Figure 7A). An increase in a CD8 T-cell signature was detected across all biopsies collected from tolinapant-treated subjects (Figure 7B), mirroring the changes seen in our in vivo preclinical models. Similarly, a broader cytotoxic cell signature was found to be elevated in 5 of 6 biopsies from treated subjects (Figure 7C). The lack of significance for the cell scores trends following multiple correction is likely due to the limited size of our dataset; hence, a follow-up study on a larger set of samples will be required to confirm the changes seen.
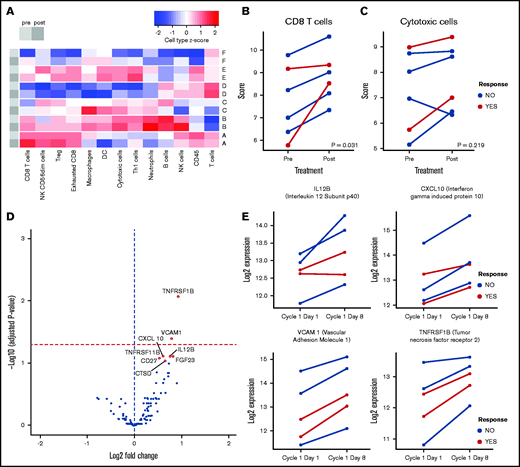
Tolinapant treatment has an immunomodulatory effect on human TCL patients. (A) Heatmap summarizing the cell type profiling scores for sets of paired PTCL biopsies from 6 patients (A-F), before (light gray) and after (dark gray) tolinapant treatment. Signature sets of gene expression associated with different cell types were scored with NanoString Advanced Analysis cell profiling module and converted to z scores. CD8 (B) and cytotoxic cells (C) scores, before vs after tolinapant treatment, in tumor biopsies of patients evaluated using NanoString Advanced Analysis modules. Responder patients are highlighted in red. All cell type scores are in log2 scale. Paired cell type scores were compared using a 2-sided Wilcoxon signed-rank test. Both P values are not significant when adjusted for multiple comparisons. (D) Volcano plot showing differentially expressed cytokines (highlighted in red circles) in plasma from patients upon treatment with tolinapant. Significant (FDR < 0.1) cytokines upregulated posttreatment are highlighted as yellow points. Horizontal red dashed line represents FDR < 0.05. (E) Log2 expression values of cytokines in plasma from 5 PTCL patients on days 1 and 8 of tolinapant cycle 1. Responder patients are highlighted in red.
Tolinapant treatment has an immunomodulatory effect on human TCL patients. (A) Heatmap summarizing the cell type profiling scores for sets of paired PTCL biopsies from 6 patients (A-F), before (light gray) and after (dark gray) tolinapant treatment. Signature sets of gene expression associated with different cell types were scored with NanoString Advanced Analysis cell profiling module and converted to z scores. CD8 (B) and cytotoxic cells (C) scores, before vs after tolinapant treatment, in tumor biopsies of patients evaluated using NanoString Advanced Analysis modules. Responder patients are highlighted in red. All cell type scores are in log2 scale. Paired cell type scores were compared using a 2-sided Wilcoxon signed-rank test. Both P values are not significant when adjusted for multiple comparisons. (D) Volcano plot showing differentially expressed cytokines (highlighted in red circles) in plasma from patients upon treatment with tolinapant. Significant (FDR < 0.1) cytokines upregulated posttreatment are highlighted as yellow points. Horizontal red dashed line represents FDR < 0.05. (E) Log2 expression values of cytokines in plasma from 5 PTCL patients on days 1 and 8 of tolinapant cycle 1. Responder patients are highlighted in red.
Of note, the biopsies from subjects who had a clinical response to tolinapant (1 complete responder and a partial responder, as detailed in Samaniego et al13 ), showed similar magnitudes of CD8 and cytotoxic signature activation compared with nonresponders, indicating that additional features are likely to be involved in defining the clinical outcome of tolinapant treatment.
To complement our transcriptomic data, plasma samples taken before and during tolinapant treatment (available for 5 of the 6 subjects) were analyzed for their cytokine profiles. A significantly increased expression of several cytokines and chemokines was detectable after tolinapant treatment (Figure 7D). Based on a false discovery rate (FDR) <0.1, a total of 8 cytokines were predicted to be significantly upregulated, including IL-12, a key regulator of T-cell differentiation and activation; IFN-γ–induced protein 10 and vascular cell adhesion molecule 1, important mediators of leukocyte recruitment; and TNF receptor 2, a known regulator of responses to IAP antagonists34 (Figure 7E; supplemental Figure 6A).
Taken together, these preliminary data support tolinapant immunomodulatory potential in the clinical setting. Future analysis of a larger cohort will be required to confirm these observations and to further characterize tolinapant effects in TCL patients.
Discussion
The generation of an antitumor immune response is a complex sequence of stepwise events that has been elegantly portrayed as the cancer-immunity cycle.35 The fine balance of inhibitory and stimulatory factors characterizing normal immune regulation is commonly altered and/or defective in the context of cancer, often leading to impaired antitumor immune responses.36,37 In the past decade, several immunotherapy approaches have been developed demonstrating the effectiveness of unleashing an anticancer immune response to curb tumor growth.38 T-cell lymphomas are a heterogeneous group of rare malignancies derived from mature T cells with limited treatment options.3 No immunotherapy treatment is currently approved for the treatment of TCL.39
Several IAP antagonists are currently in clinical development, but no single-agent clinical activity has previously been described for this class of compounds.25 Interestingly, the IAP antagonist tolinapant, which is currently being evaluated in a phase 2 study, has demonstrated preliminary evidence of single-agent activity in relapsed/refractory PTCL and cutaneous T-cell lymphoma.13,14 To investigate the mechanism of action behind the single-agent activity observed in the clinic with tolinapant in TCL, we have used a comprehensive translational approach via integration of in vitro and in vivo preclinical models along with human patient data from an ongoing clinical trial (NCT02503423).
In this manuscript, we demonstrate that tolinapant acts as an efficacious immunomodulatory molecule in TCL preclinical models. We found that single-agent tolinapant induces long and complete tumor regressions in a murine syngeneic model of TCL in the presence of an intact immune system. Importantly, mice previously cured by tolinapant were protected when rechallenged with fresh tumor cells, clearly implicating a role of adaptive immunity in generating a durable antitumor immune response following tolinapant treatment. Interestingly, the regressions observed in this syngeneic model mirrored the single-agent effects noted in the clinic, with some PTCL patients experiencing long-term complete responses (on treatment of more than 1 year).13 Taken together, these data demonstrate a clear immunomodulatory role for tolinapant as a single agent and complement recent studies suggesting a role of tolinapant in immune cell modulation in combination settings.31,40
Our studies unveil the complex nature of tolinapant’s mechanism of action, simultaneously acting in a multimodal fashion on several components of the antitumor immune response. First, we show that tolinapant has a direct effect on T cells by increasing their antitumor killing capabilities as shown by our in vitro coculture systems based on primary leukocytes from healthy donors. This is likely from the action of IAP antagonists on lymphocytes via the increase of TNF-α and IFN-γ secretion and by stimulation of the noncanonical nuclear factor-κB pathway, which is a key regulator of T-cell costimulatory pathways.17 Second, tolinapant is sensitizing tumor cells to cytokine-mediated cell death, converting survival stimuli into death-inducing signals.26,41 Cytokine-mediated cell death in the presence of IAP antagonists may include cell-to-cell contact-dependent engagement of the TNF superfamily (TNF/TRAIL/FAS) and their receptors, a mechanism implicated in T cell–dependent bystander killing.42
Third, we show that tolinapant treatment can promote T-cell infiltration into the tumor. In preclinical models, a proinflammatory tumor microenvironment with an increased CD8+ T-cell infiltration was detected after tolinapant treatment. This is the first evidence that an IAP antagonist can increase tumor T-cell infiltration as a single agent in preclinical models. An important part of our study is the translation of our preclinical findings to biopsies from PTCL subjects. Transcriptomic analysis of PTCL biopsies pre- and posttreatment showed an increased CD8+ T-cell signature after tolinapant administration, indicating that the observed mechanism of action of tolinapant in preclinical models could be translatable to human TCL. Moreover, the increased CD8 and cytotoxic signature observed in the 2 biopsies collected from subjects who responded to tolinapant supports our preclinical findings, indicating that an antitumor immune response could be necessary to mediate clinical responses to tolinapant. This finding represents the first evidence that an IAP antagonist can be used to increase cytotoxic T-cell infiltration in human tumors. Should these preliminary clinical observations be confirmed, we expect that novel opportunities could be envisaged for the use of tolinapant in the treatment of immune-cell excluded tumors, known to be unresponsive to checkpoint inhibitor treatment.38,43
In addition to its effects on the adaptive immune system, we show that tolinapant is also acting on the innate arm, an effect of IAP antagonists which has been previously reported.18 Our data show that in vitro tolinapant has a direct effect on monocytes, leading to increased activation and M1 polarization. In vivo tolinapant treatment leads to a strong upregulation of a myeloid signature in 2 xenograft models of TCL (Figure 4; supplemental Figure 4). In addition to these direct effects, our studies also uncover a novel indirect mechanism by which tolinapant can boost myeloid activation. Various forms of ICD, such as necroptosis, have been described.10,44,45 Necroptosis leads to rapid plasma membrane permeabilization and release of DAMPs.46 DAMPs are specifically recognized by myeloid cells, initiating signaling pathways that are required for myeloid activation leading to increased antigen presentation and/or phagocytosis.27,30,47 Our in vivo rechallenge and vaccination experiments demonstrate that tolinapant can activate immunogenic forms of cell death in TCL cells, leading to release of DAMPs and modulation of the tumor microenvironment, thus opening the way for the development of an effective adaptive antitumor immune response.
Promising preliminary reports from the ongoing phase 2 clinical data indicate complete and partial responses obtained with tolinapant.13 To our knowledge, single-agent activity with an IAP antagonist has not been reported in other indications. This study seeks to identify the underlying mechanisms for this efficacy and expand our understanding of the biologic activities of IAPs. The dual effect of tolinapant, both as an immunomodulatory molecule and cell death sensitizer, may be crucial for its single agent activity in TCL.
Overall, we have uncovered novel mechanisms of action of IAP antagonist therapy demonstrating its activity via multiple steps in the cancer immunity cycle. We envisage that the ability of tolinapant to act on both the innate and the adaptive arm of the immune system can be exploited as a single-agent cancer therapy to remodel the tumor immune microenvironment, facilitating the establishment of an antitumor immune response. The increased understanding of the biological mechanism underlying tolinapant’s single-agent activity in the clinic will be key to guiding patient selection strategies and developing future combination options that could greatly benefit TCL patients.
Acknowledgments
The authors thank all the patients who participated in and contributed samples to the study and acknowledge scientific discussions with many Astex colleagues. The visual abstract linked to this article was created with BioRender.com.
This study was fully supported by research funding from Astex Pharmaceuticals.
Authorship
Contribution: N.F., G.W., C.G., and M.J.S., conceived and designed the study and interpreted the results; N.F., G.W., and C.G. conducted the experiments and performed data analysis; J.M. and T.S. helped with experimental work and interpretation of results; M.P.D., N.F., and H.S. performed bioinformatics analyses; S.J. assisted with obtaining, processing, and data generation from patient samples; R.F. and H.K. designed and managed the clinical trial; N.F. wrote and G.W., M.J.S., and J.L. edited the manuscript; and all authors discussed the results and commented on the manuscript.
Conflict-of-interest disclosure: All authors are employees of Astex Pharmaceuticals.
The current affiliation for R.F. is Pharma Research & Early Development (pRED) Oncology, Roche, Zurich, Switzerland.
Correspondence: Martin Sims, Astex Pharmaceuticals, 436 Cambridge Science Park, Milton Rd, Cambridge, UK; e-mail: martin.sims@astx.com; and Nicola Ferrari, Astex Pharmaceuticals, Cambridge, UK; e-mail: nicola.ferrari@astx.com.