Key Points
Interferon-γ–related chemokines are highly expressed in unbiased analysis of plasma proteins in children with HLH vs sepsis or SIRS.
Plasma protein classifiers that include CXCL9 and IL-6 differentiate children with HLH vs sepsis or SIRS.

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome characterized by pathologic immune activation in which prompt recognition and initiation of immune suppression is essential for survival. Children with HLH have many overlapping clinical features with critically ill children with sepsis and systemic inflammatory response syndrome (SIRS) in whom alternative therapies are indicated. To determine whether plasma biomarkers could differentiate HLH from other inflammatory conditions and to better define a core inflammatory signature of HLH, concentrations of inflammatory plasma proteins were compared in 40 patients with HLH to 47 pediatric patients with severe sepsis or SIRS. Fifteen of 135 analytes were significantly different in HLH plasma compared with SIRS/sepsis, including increased interferon-γ (IFN-γ)–regulated chemokines CXCL9, CXCL10, and CXCL11. Furthermore, a 2-analyte plasma protein classifier including CXCL9 and interleukin-6 was able to differentiate HLH from SIRS/sepsis. Gene expression in CD8+ T cells and activated monocytes from blood were also enriched for IFN-γ pathway signatures in peripheral blood cells from patients with HLH compared with SIRS/sepsis. This study identifies differential expression of inflammatory proteins as a diagnostic strategy to identify critically ill children with HLH, and comprehensive unbiased analysis of inflammatory plasma proteins and global gene expression demonstrates that IFN-γ signaling is uniquely elevated in HLH. In addition to demonstrating the ability of diagnostic criteria for HLH and sepsis or SIRS to identify groups with distinct inflammatory patterns, results from this study support the potential for prospective evaluation of inflammatory biomarkers to aid in diagnosis of and optimizing therapeutic strategies for children with distinctive hyperinflammatory syndromes.
Introduction
Hemophagocytic lymphohistiocytosis (HLH), severe sepsis, and persistent systemic inflammatory response syndrome (SIRS) are all conditions defined by excessive immune activation that may progress rapidly and are associated high risk of death without early introduction of appropriate therapy. HLH is functionally defined by the HLH 2004 criteria that identifies patients with extreme inflammation caused by inherited defects in cytotoxic effector T and natural killer cells and/or persistent pathologic immune activation associated with infections, malignancy, or autoimmune disease1-3. SIRS is an activated state of systemic inflammation meeting slightly different clinical criteria, but without an identifiable infectious cause. Alternatively, symptoms of SIRS in response to a confirmed infection is defined as sepsis in pediatrics.4,5 Urgent introduction of antimicrobial therapy is essential for children with severe sepsis, where mortality rates increase by 7% every hour that appropriate antimicrobial therapy is delayed. In contrast, early initiation of immune suppressive immunochemotherapy is deemed imperative for children with HLH,6,7 whereas this may seem excessive or contraindicated for children with persistent SIRS or sepsis.
Clinically, HLH, sepsis, and persistent SIRS share many features including fever, hemodynamic compromise, and signs of organ damage or failure. The diagnostic criteria for HLH, severe sepsis and SIRS are largely derived from committee consensus with limited biological validation, and it has been argued that in some cases, HLH, severe sepsis, and SIRS may not be biologically distinct; rather, these conditions may fall along a continuum of inflammation with the specific syndrome influenced by bias of the diagnosing physician.8 In this study, we tested the ability of unbiased comparison of an extensive inflammatory plasma protein panel and gene expression of CD3+8+ T cells and CD68+ monocytes from peripheral blood to distinguish children diagnosed with active HLH (as defined by the HLH2004 criteria1 ) from children with severe sepsis or SIRS (as defined by the 2005 International Pediatric Sepsis Definitions Conference4,5,9 ) to determine whether biological differences exist in mechanisms of inflammation between these clinically defined groups.
Methods
Study patients and samples
Blood samples were collected, processed, and frozen from 111 patients under protocols approved by the Baylor College of Medicine and Cincinnati Children’s Hospital Institutional Review Boards. Ten cases were from subjects enrolled on the HIT-HLH trial (NCT01104025) who consented to correlative biology studies. The study was conducted in accordance with the Declaration of Helsinki. This series included 40 patients with active HLH, 26 with severe sepsis, and 21 with SIRS (Tables 1-2; supplemental File 1). HLH patients were defined by the HLH 2004 diagnostic criteria1 (supplemental Table 1A) with active disease at the time of blood collection. Patients who received more than 1 dose of etoposide, had prolonged steroid use (>10 mg/m2 per day dexamethasone equivalent for any period of time or any dose of corticosteroids for >3 months), or received alemtuzumab or anti–thymocyte globulin before sample draw were excluded. None of the sepsis or SIRS patients received immune suppression before collection of blood specimens. SIRS and severe sepsis were defined by 2005 International Pediatric Sepsis Definitions Consensus Conference.4,5,9 (supplemental Table 1B-C). Control subjects were obtained from children without known infections or immune disorders. Blood samples were processed within 48 hours, and plasma was separated into aliquots, stored at −80°C, and underwent a maximum of 1 cycle of freezing and thawing.
Plasma protein analysis
Plasma protein levels of 135 analytes representing cytokines, chemokines, and growth factors were determined using Milliplex MAP antibody panels (Millipore, Burlington, MA) for the MagPix instrument (Luminex, Austin, TX) outlined in supplemental Table 2. Ferritin and interleukin-18 (IL-18) were analyzed in independent MagPix assays. Antibodies for some proteins are included in more than 1 kit but are analyzed as independent analytes in this study and represented with consistent designation throughout the study (eg, protein[1], protein[2], protein[3]). The concentration of each analyte was measured by comparing to the protein standards. A logarithmic (base 2) transformation was applied to the sample concentrations before quality control. Sample-sample correlations (Spearman correlation) and overall distribution of log concentrations (box plots) were used to assess the quality of the dataset. No samples were rejected based on these criteria. The assays were organized so that the relative proportion of each class of sample was preserved as much as possible from plate to plate to minimize batch effects.
Before any analysis, the plasma dataset was randomly partitioned into a training and validation cohort such that each cohort had a similar breakdown with regard to clinical characteristics of interest. A Pearson’s χ2 test was used to test whether the patient diagnosis/clinical parameters and the grouping of the samples were not statistically significant different from each other (P < .05; Tables 1-2). For both the plasma biomarker comparisons and the development of the diagnostic classifier, analytes were first discovered in the training set and then independently validated in the validation set.
Plasma biomarker analysis
Hierarchical clustering and principal component analysis were performed using R version 3.6.1 (www.r-project.org). Hierarchical clustering was performed using the hclust package with Pearson correlation and the complete agglomeration method. Principal component analysis was then performed using all features.
A univariate parametric t test was used to compare clinical characteristics of interest in a training cohort with a multivariate permutation test with confidence level of false discovery rate (FDR) assessment at 80% and the maximum allowed proportion of false-positive proteins at 0.1. The resulting analytes were considered biomarkers of interest and tested in the validation cohort using a similar methodology. Additional pairwise comparisons of selected analytes of interest (eg, interferon-γ [IFN-γ], CXCL9, CXCL10, CXCL11) were performed using the Mann-Whitney-Wilcoxon test because they did not satisfy normality assumption based on the Shapiro-Wilk test.
Diagnostic classifier development
A classifier, or combination of protein concentrations, was developed to distinguish the HLH plasma protein profile from SIRS/severe sepsis. The model was built using the training cohort, and then used to subsequently classify new samples in the validation cohort. Duplicate features and features with more than 85% missing values were filtered out before model building. Feature selection criteria was determined using a greedy pairs algorithm to select 5 pairs of analytes (or 10 analytes total). A compound covariate predictor classifier was then chosen from among 8 different classifiers (compound covariate predictor, diagonal linear discriminant analysis, 1-nearest neighbor, 3-nearest neighbors, support vector machine, bayesian compound covariate predictor, and nearest centroid) based on overall classification accuracy in the training cohort. To construct a classifier with a minimal number of analytes, a second classifier using only CXCL9 and IL-6, which were both present in the first classifier, was subsequently developed using the training set and independently tested on the validation set using a similar compound covariate predictor. Leave-one-out cross-validation was used to compute misclassification rate in the training set. Receiver operating characteristic (ROC) curves were then generated, and the areas under the ROC curves (AUC) were calculated for both classifiers within the training and validation sets.
Gene expression analysis
Peripheral blood mononuclear cells were sorted for CD3 (BD 555335), CD4 (BD 550631), CD8 (BD 555634), and CD68 (Biolegend 333808) using the BD Aria Fusion (BD Bioscience, Franklin Lakes, NJ). cDNA was prepared from CD3+8+ and CD3−68+ populations with the Nugen Ovation Pico WTA System V2 (3302-12) (Nugen, Redwood City, CA) (supplemental Figure 1). Gene expression data were generated using Affymetrix GeneChip Human Transcriptome Array 2.0 (Affymetrix, Santa Clara, CA) at Genome Explorations (Memphis, TN). This series included a CD3+8+ subpopulation cohort with 12 samples with HLH and 10 with severe sepsis. The CD3−68+ subpopulation cohort had 12 samples with HLH and 8 with severe sepsis. Data are available at the Gene Expression Omnibus website (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150707).
Gene set enrichment analysis
We performed gene set enrichment analysis (GSEA) for The Molecular Signatures Database (MSigDB) hallmark gene set collection in both the CD3+8+ and the CD3−68+ subpopulation cohorts. A preranked GSEA was run with The Broad Institute’s GSEA software, where genes within the gene expression cohorts (Affymetrix) were ranked by multiplying the sign of the log fold change by the negative log of the unadjusted P value. To collapse the ranked list to gene symbols, the higher ranked feature, if a feature had duplicate gene symbols, was taken.
The IFN-γ gene signature from a recent publication that examined gene expression in the tumor microenvironment and was able to validate their signature across 9 different cancer cohorts was used to investigate the enrichment of genes specifically related to IFN-γ between HLH and sepsis and HLH and control.10
Results
Plasma proteomics patient characteristics
Of the 40 pediatric HLH subjects, 9 had biallelic gene mutations known to cause HLH (or monoallelic X-linked SH2D1A), 4 had single allele variants associated with HLH, and 11 had variants of uncertain significance in genes associated with immune functions potentially associated with immune dysregulation and HLH susceptibility.11 Three HLH patients had associated autoimmune disease (systemic juvenile idiopathic arthritis [n = 2]; drug reaction with eosinophilia and systemic symptoms [n = 1]). Fifteen had infections at the time of diagnosis, including 7 with highly elevated Epstein-Barr virus levels. Three had malignancy-associated HLH: infant acute lymphoblastic leukemia on maintenance therapy, subcutaneous panniculitis-like T-cell lymphoma and anaplastic large cell lymphoma with active malignancy. In 1 patient with Griscelli syndrome, HLH preceded development of peripheral T-cell lymphoma, so this case was considered primary (Table 2; supplemental File 1A). Therefore, 85% (34 of 40) were defined as having primary HLH and 15% (6 of 40) as secondary, with primary indicating HLH in the absence of autoimmune disease or cancer and secondary associated with autoimmune disease or cancer.12 Notably, analysis of the HLH plasma proteome did not identify any differentially expressed analytes with supervised comparison of primary vs secondary HLH or between familial HLH (cases with identified HLH-associated gene defects) and nonfamilial (cases without HLH-associated gene defects identified). Furthermore, unsupervised clustering demonstrates lack of uniform grouping of the HLH cohort (Figure 1A). One case (HLH33) with HLH-associated RAB27A mutations and T-cell lymphoma was classified as primary. There were 26 subjects in the severe sepsis cohort and 21 subjects in the SIRS cohort. Clinical features are outlined in supplemental File 1B.
![Unsupervised analysis does not distinguish HLH from sepsis/SIRS compared with pediatric controls. (A) Heat map displaying unsupervised clustering of all plasma analytes assayed from noninflammatory pediatric controls, HLH, sepsis, and SIRS. Colors representing specific diagnostic groups/subgroups are included in the legend. Cases of secondary HLH (systemic juvenile idiopathic arthritis [JIA], drug reaction with eosinophilia syndrome [DRESS], and cancer) are indicated. (B) These datasets are also presented using principal component analysis. The control cases clearly separate, whereas HLH, severe sepsis, and SIRS have overlapping inflammatory plasma profiles.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/17/10.1182_bloodadvances.2021004287/2/m_advancesadv2021004287f1.png?Expires=1766075238&Signature=zyR~bakvxhIGaXYngGO1NEuC0GUEbYZB8rmd7neHJGmggqjEDEGmC1jyBB~1qu-OkL1FSk66veh-asVYpW~SovWi86RDXIHCFog51ahnibfUzTGXpBmazZ539q88eJDxgcmhrlRwixE7j9BtWXwDPzLO4UwkCToYgmZ6oEhQu2O4SseHbHF8wUwGXBIA0yAs8DqmhFHHtHNM7VTk1OzzGgEvgknd7sMPZwTQno7JKSL4YHkGCgnlXxgQHSdKeAkiQ4zL-4BwUHEY2aPPHHoktVNIQmLRVElsR9G1i4DEdmeQV8cZjisn0aKvrsy8nATnQvgpRWQtoI3RAkIOmFPg2Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Unsupervised analysis does not distinguish HLH from sepsis/SIRS compared with pediatric controls. (A) Heat map displaying unsupervised clustering of all plasma analytes assayed from noninflammatory pediatric controls, HLH, sepsis, and SIRS. Colors representing specific diagnostic groups/subgroups are included in the legend. Cases of secondary HLH (systemic juvenile idiopathic arthritis [JIA], drug reaction with eosinophilia syndrome [DRESS], and cancer) are indicated. (B) These datasets are also presented using principal component analysis. The control cases clearly separate, whereas HLH, severe sepsis, and SIRS have overlapping inflammatory plasma profiles.
Unsupervised analysis does not distinguish HLH from sepsis/SIRS compared with pediatric controls. (A) Heat map displaying unsupervised clustering of all plasma analytes assayed from noninflammatory pediatric controls, HLH, sepsis, and SIRS. Colors representing specific diagnostic groups/subgroups are included in the legend. Cases of secondary HLH (systemic juvenile idiopathic arthritis [JIA], drug reaction with eosinophilia syndrome [DRESS], and cancer) are indicated. (B) These datasets are also presented using principal component analysis. The control cases clearly separate, whereas HLH, severe sepsis, and SIRS have overlapping inflammatory plasma profiles.
Global activation of inflammation in HLH, sepsis, and SIRS vs healthy controls
Consistent with overlapping clinical features of fever, organ failure, and infection, patients with HLH, severe sepsis, and SIRS all had increased concentrations of many inflammatory plasma proteins: 40% (54 of 135) of the analytes tested were significantly different between HLH plasma and that of healthy controls (supplemental Figure 2A); similarly, 46% (62 of 135) of the analytes were significantly different between sepsis or SIRS (sepsis/SIRS) plasma and that of healthy controls (supplemental Figure 2B). Unsupervised clustering was able to differentiate healthy control patients from those with active inflammatory disorders but was not able to consistently separate HLH, sepsis, and SIRS (Figure 1A). Similarly, principal component analysis separated control plasma proteome where sepsis overlapped with both SIRS and HLH datasets (Figure 1B).
IFN-γ responsive proteins increased in HLH vs sepsis/SIRS.
To determine whether there were differences between HLH and other inflammatory conditions, the HLH plasma dataset was compared with severe sepsis or SIRS cases. Fifteen analytes were significantly different between HLH and either sepsis or SIRS (FDR = 0.1), and there were no significant differences in concentration of any plasma cytokine/chemokines between SIRS and severe sepsis cases in this study.
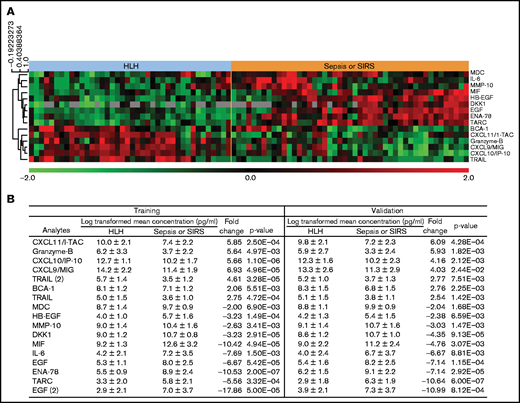
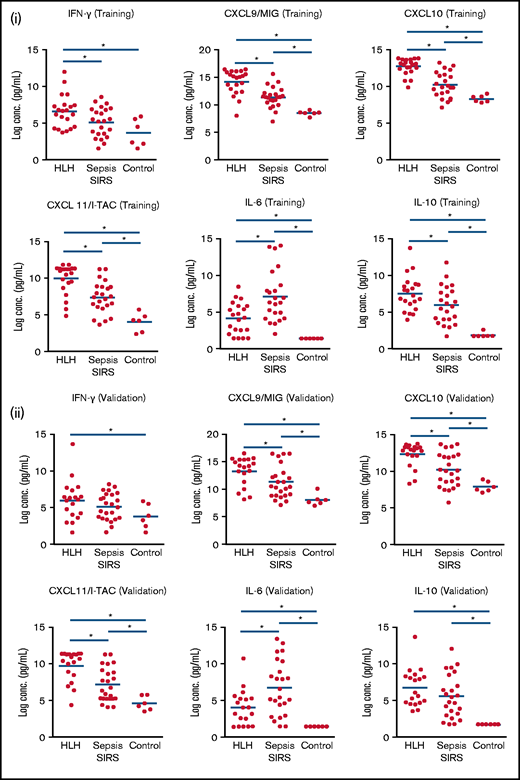
Among the most highly differentially expressed proteins from this unbiased analysis in HLH were IFN-γ–induced proteins and CXCR3 ligands: CXCL9, CXCL10, and CXCL1113 (Figure 2). Pairwise comparison of HLH vs sepsis/SIRS demonstrated elevation of IFN-γ in HLH and sepsis/SIRS compared with noninflammatory controls (Figure 3). However, CXCL9, CXCL10, and CXCL11 were significantly elevated in both the training and validation HLH plasma cohorts compared with sepsis/SIRS. Relatively decreased expression of IL-6 and increased expression of IL-10 have been reported between HLH and patients without HLH with infections.14 In this study, IL-6 was elevated in both HLH and sepsis/SIRS compared with controls but was significantly less elevated in HLH compared with sepsis/SIRS. IL-10 was elevated in HLH and sepsis/SIRS, with no significant difference between the hyperinflammatory group validation sets.
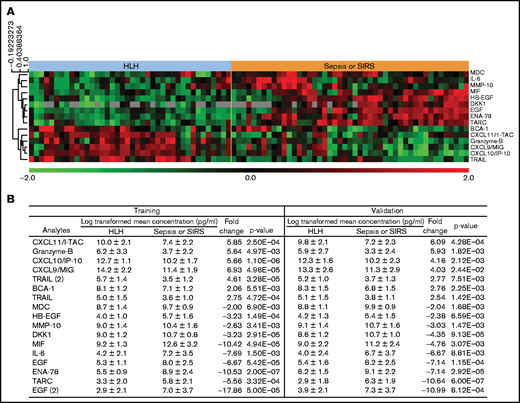
Plasma analytes with significant differences between HLH and sepsis/SIRS. (A) Heatmap demonstrates relative concentration of analytes with significant differential expression (FDR < 0.1) revealed by supervised comparison of HLH and SIRS/sepsis datasets. All subjects in this study are shown with heatmap reflecting relative concentrations of analytes with significant differential expression between HLH and sepsis or SIRS in both training and validation cohorts. (B) Subjects were split into training and validation cohorts, without reference to cytokine values, and analyzed separately. Values of analytes with differential expression between HLH and SIRS/sepsis datasets are displayed for training and validation cohorts.
Plasma analytes with significant differences between HLH and sepsis/SIRS. (A) Heatmap demonstrates relative concentration of analytes with significant differential expression (FDR < 0.1) revealed by supervised comparison of HLH and SIRS/sepsis datasets. All subjects in this study are shown with heatmap reflecting relative concentrations of analytes with significant differential expression between HLH and sepsis or SIRS in both training and validation cohorts. (B) Subjects were split into training and validation cohorts, without reference to cytokine values, and analyzed separately. Values of analytes with differential expression between HLH and SIRS/sepsis datasets are displayed for training and validation cohorts.
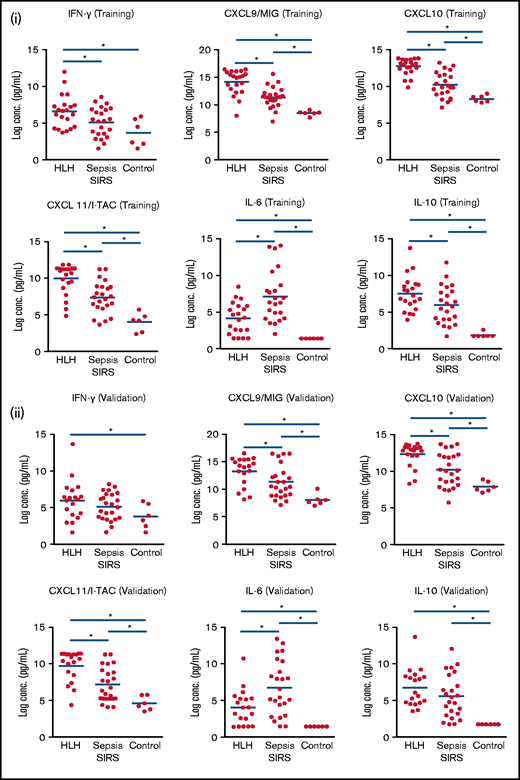
IFN-γ–inducible plasma proteins are significantly increased in HLH. Boxplots demonstrate comparisons of specific analyzes in HLH, SIRS/sepsis, and control groups in training (i) and validation (ii) cohorts. Bars with asterisk indicate groups with statistically significant differential expression evaluated using a Mann-Whitney-Wilcoxon test (P < .05). IFN-γ–inducible plasma proteins CXCL9, CXCL10, and CXCL11 are significantly increased in HLH compared with sepsis in training and validation cohorts. IL-6 is relatively increased in SIRS/sepsis compared with HLH, and there is no significant difference in IL-10 concentrations in HLH and SIRS/sepsis in the validation cohort in this study.
IFN-γ–inducible plasma proteins are significantly increased in HLH. Boxplots demonstrate comparisons of specific analyzes in HLH, SIRS/sepsis, and control groups in training (i) and validation (ii) cohorts. Bars with asterisk indicate groups with statistically significant differential expression evaluated using a Mann-Whitney-Wilcoxon test (P < .05). IFN-γ–inducible plasma proteins CXCL9, CXCL10, and CXCL11 are significantly increased in HLH compared with sepsis in training and validation cohorts. IL-6 is relatively increased in SIRS/sepsis compared with HLH, and there is no significant difference in IL-10 concentrations in HLH and SIRS/sepsis in the validation cohort in this study.
Gene expression enriched for IFN-γ signaling in HLH peripheral blood populations.
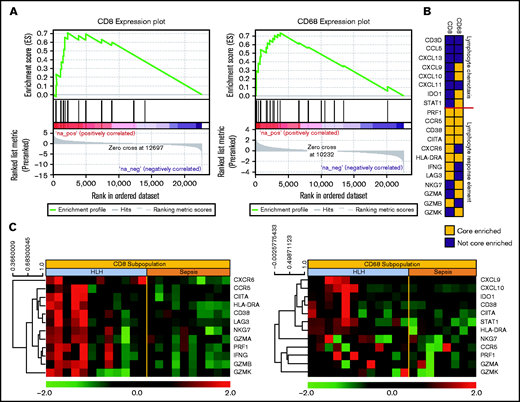
To further define mechanisms underlying differential plasma cytokines, we evaluated gene expression profiles of effector cytotoxic T cells (CD3+CD8+) and activated CD68+ monocytes from peripheral blood of patients with newly diagnosed HLH and severe sepsis. GSEA was used to investigate the differences in biological pathways related to HLH and sepsis. Using the refined hallmark gene set collection curated by MSigDB, E2F Targets and G2M Checkpoint pathways were significantly enriched in HLH compared with sepsis CD3+8+ cells with an FDR q-value of 0.05 or less. Notably, the IFN-γ response was enriched in the top 10 pathways in HLH CD3+8+ cells with an FDR q-value of 0.1 or less. The top pathways that were significantly enriched with an FDR q-value of 0.05 or less in HLH compared with Sepsis in CD68+ monocyte subpopulations also included the IFN-γ response (Figure 4A).
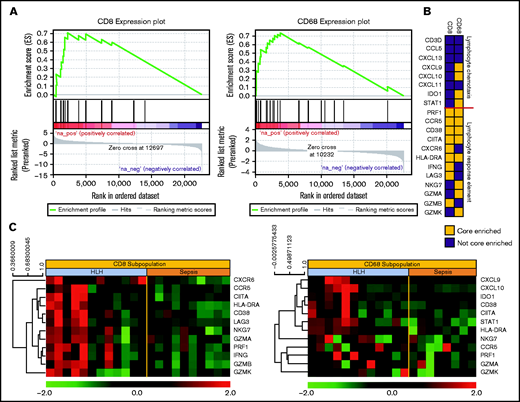
Gene expression signatures validate plasma classifier and point to distinctive T-cell and IFN-γ–driven etiology of HLH. (A) Gene set enrichment plot showing the enrichment score of the ranked gene list in the IFN–γ response signature of CD8+ T cells and CD68+ monocytes from subjects with HLH and severe sepsis. (B) Genes included in the leading edge analysis of the IFN-γ response signature (A) are presented graphically. The leading edge consists of the subset of genes in the signature where the running sum of the enrichment score reaches its maximum deviation from zero in panel A. The color describes whether the gene is (yellow) or is not (blue) present in leading-edge genes in the CD8 or CD68 datasets. (C) Relative gene expression of the leading edge genes that are significantly enriched in the INF-γ response signature from the CD8 and CD68 subpopulations are presented in a heatmap, with each column representing cell-specific transcriptome from a single subject.
Gene expression signatures validate plasma classifier and point to distinctive T-cell and IFN-γ–driven etiology of HLH. (A) Gene set enrichment plot showing the enrichment score of the ranked gene list in the IFN–γ response signature of CD8+ T cells and CD68+ monocytes from subjects with HLH and severe sepsis. (B) Genes included in the leading edge analysis of the IFN-γ response signature (A) are presented graphically. The leading edge consists of the subset of genes in the signature where the running sum of the enrichment score reaches its maximum deviation from zero in panel A. The color describes whether the gene is (yellow) or is not (blue) present in leading-edge genes in the CD8 or CD68 datasets. (C) Relative gene expression of the leading edge genes that are significantly enriched in the INF-γ response signature from the CD8 and CD68 subpopulations are presented in a heatmap, with each column representing cell-specific transcriptome from a single subject.
To validate the IFN-γ response signature result, we further analyzed the gene signature in CD8+ cytotoxic T-cell lymphocytes and CD68+ monocytes in HLH or sepsis using an independent IFN-γ signature.10 Because not all the members of a gene set are involved in a biological process, it will be important to identify the core members of the IFN-γ response signature that contribute to the enrichment score. Thus, we examined the genes comprising the leading-edge subsets of the signature identified enrichment for lymphocyte response in cytotoxic T-cell lymphocytes and for response elements in the monocytes (Figure 4B). The leading-edge subset can be interpreted as the group of genes that most accounts for a given gene set’s enrichment signal. Identification of the leading-edge subset can reveal biologically significant genes within a gene set.15 These gene expression patterns support a model of hyperactivated, proliferating CD8+ cytotoxic T cells that produce IFN-γ and induce expression IFN-γ–responsive genes in activated peripheral blood monocytes. Data files for gene expression studies are available in GSE150707.
Diagnostic classifier predicts HLH vs SIRS/sepsis.
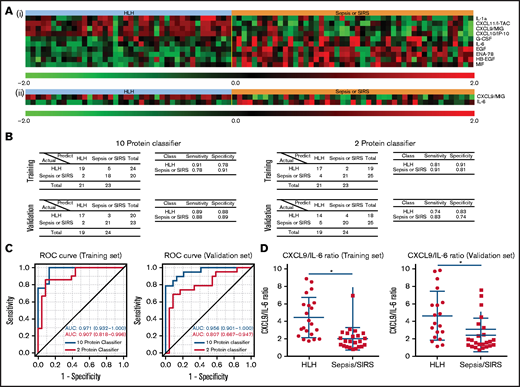
To test whether circulating proteins can be used to distinguish HLH cases from SIRS/severe sepsis cases, we developed 2 plasma protein classifiers. For the first classifier, we adopted an unbiased approach by identifying the most informative features for the classification in a training set. The resultant classifier consisted of 10 proteins with increased CXCL9, CXCL10, CXCL11, and IL-1a with decreased IL-6, heparin-binding epidermal growth factor (EGF)-like growth factor (HB-EGF), epithelial-derived neutrophil-activating peptide 78 (also known as CXCL5), macrophage migration inhibition factor, and EGF (Figure 5A). When applying to an independent validation cohort, the classifier was able to differentiate HLH patient plasma from SIRS/severe sepsis patient plasma with 89% specificity and 88% sensitivity (Figure 5B). Because clinical assays of IFN-γ signaling proteins and IL-6 are clinically available, we specifically selected only CXCL9 and IL-6 to construct the second classifier in the training cohort (Figure 5A).14 When applied to an independent validation cohort, the classifier was able to differentiate HLH patient plasma from SIRS/severe sepsis patient plasma with 74% specificity and 83% sensitivity (Figure 5B). To compare the performances of the 2 classifiers, we performed ROC curve analysis (Figure 5C). The AUC of the 10-protein classifier in the training set was 0.971 and 0.956 in the validation set, respectively. In comparison, AUC of the 2-protein classifier decreased to 0.907 and 0.807 in the training and validation sets, respectively. Although the CXCL9 and IL-6 classifier performed well, the other analytes in the 10-protein classifier have additional discriminatory value. Additionally, CXCL9/IL-6 ratios were significantly elevated in HLH compared with SIRS/sepsis and discriminated HLH from SIRS/sepsis (Figure 5D; supplemental Figure 4).
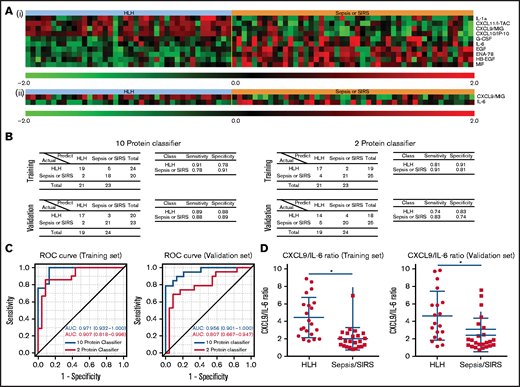
Protein classifiers to distinguish HLH from sepsis/SIRS. (A) Full (i) and reduced (ii) protein classifiers were developed in a training cohort using a compound covariate predictor to differentiate HLH from SIRS/sepsis and subsequently validated using an independent cohort. The heatmap shows the features used in those classifiers in the combined training and validation sets. (B) Confusion matrices, sensitivities, and specificities of the 2 classifiers in both the training and validation cohorts. (C) Receiver operating curves of both full and reduced classifiers in the training and validation sets. The AUC was calculated for each classifier as an overall diagnostic accuracy.
Protein classifiers to distinguish HLH from sepsis/SIRS. (A) Full (i) and reduced (ii) protein classifiers were developed in a training cohort using a compound covariate predictor to differentiate HLH from SIRS/sepsis and subsequently validated using an independent cohort. The heatmap shows the features used in those classifiers in the combined training and validation sets. (B) Confusion matrices, sensitivities, and specificities of the 2 classifiers in both the training and validation cohorts. (C) Receiver operating curves of both full and reduced classifiers in the training and validation sets. The AUC was calculated for each classifier as an overall diagnostic accuracy.
HLH cohort comparisons.
We did not detect any significantly different analytes comparing primary and secondary HLH as defined above; similarly, there were no significantly different analytes comparing cases with proven inherited HLH-causing gene defects (familial) and those without proven HLH gene defects (supplemental Figure 5). However, because of the relatively small sample sizes used in these comparisons, it is likely that the analyses do not have enough statistical power to detect the differential expressed proteins. Future larger studies are needed to confirm these results.
Several previous studies have also identified increased ferritin and IL-18 in patients with active HLH and macrophage activation syndrome.16-18 Analytes for these plasma proteins were not available to be included in multiplex panels, and volume required for enzyme-linked immunosorbent assay limited inclusion in global proteome analysis or classifier development. However, pairwise comparison with a representative set of plasma samples identified significantly higher expression of both ferritin and IL-18 in HLH compared with severe sepsis and SIRS (supplemental Figure 6).
Discussion
Children with HLH continue to have very high risk of death, and successful treatment requires prompt diagnosis and initiation of immune suppressive therapy. However, diagnosis of HLH is confounded by overlapping features with other inflammatory disorders. Furthermore, HLH is a syndrome of pathologic hyperinflammation with multiple inherited and acquired causes. Previous studies have described elevated inflammatory markers in children with active HLH but have not compared HLH with other inflammatory conditions or have assayed only limited numbers of analytes in narrow populations.19,20 The results from this study demonstrate that as a group, children meeting criteria for HLH 2004 can indeed be distinguished from other critically ill children in the intensive care unit with SIRS or severe sepsis, and this pattern points to a unique T-cell and IFN-γ–mediated etiology for HLH.
Other studies have investigated plasma cytokines or gene expression in HLH with various methods and populations. We previously reported that highly elevated ferritin (>10 000 mg/dL) differentiated HLH patients from other patients with ferritin > 500 mg/dL in an institutional retrospective cohort with >90% sensitivity and specificity.16 Extended analysis with additional institutions suggested highly elevated ferritin (>2000 mg/dL) was sensitive for pediatric HLH with approximately 70% sensitivity and specificity.21 These studies were limited by patients in whom ferritin levels were obtained, but demonstrate potential utility of measuring amplitude of specific acute phase reactants and cytokines/chemokines to differentiate HLH from other conditions. Tang and colleagues20,22,23 studied children with fever and found increased IL-6, IL-10, and IFN-γ in HLH, but relatively greater increase in IFN-γ and IL-10 in patients with HLH compared with fever of other causes including septic shock. Among children with hemophagocytic syndromes, increases in IFN-γ and IFN-γ–induced chemokines are higher in patients with HLH/macrophage activation syndrome (MAS) compared to those with active juvenile idiopathic arthritis without MAS24-26 and also in lymphoma-associated HLH.27 Chen et al28 reported lower IL-4 and IFN-γ in primary HLH (defined by familial HLH mutations associated with cytotoxic function) compared with secondary (mostly Epstein-Barr virus associated).
This study was unique with respect to the scope of inflammatory proteins analyzed and overall design, using unbiased methodology and correction for multiple comparisons. The vast difference between healthy control proteome and that of critically ill children with HLH or SIRS or severe sepsis (>40% of inflammatory proteins differentially expressed; supplemental Figures 2 and 3) demonstrates the potential impact of other highly inflamed comparison groups on identification of HLH-associated proteins. Although IFN-γ was not identified as differentially expressed in this study compared within the entire dataset and correcting for multiple comparisons, pairwise comparison demonstrated increased IFN-γ expression compared with sepsis/SIRS in the training series and a trend toward increased expression in the validation cohort (Figure 3). However, the known function of IFN-γ as an activator of macrophages appears to be a highly localized action within tissues and would not be expected to be directly detectable in plasma. However, the IFN-γ−induced chemokines, CXCL9, CXCL10, and CXCL11, which act at a distance to call immune cells into inflamed tissues,29 were readily detectable in plasma and highly dynamic in our cohort. These chemokines were elevated in nearly all patients with HLH and readily differentiated HLH from sepsis and SIRS in this study. Although inflammation is globally activated in each of these conditions, relative to healthy control children, in HLH, the cytokine storm is dominated by IFN-γ signaling. These findings are consistent with a critical role for IFN-γ signaling in mouse models of primary and secondary HLH30,31 and with studies that have identified IFN-γ and IFN-γ–responsive proteins to reflect disease activity in HLH and MAS in both mouse models and human patients with hemophagocytic syndromes.14,20,24,28,32,33 IFN-γ is expressed in a highly directional fashion, with much of the cytokine acting locally on interacting cells.34 Therefore, plasma IFN-γ–responsive proteins CXCL9, CXCL10, and CXCL11 may be more sensitive reflection of total IFN-γ levels (tissue-bound and free) than plasma IFN-γ and have been shown to correlate with disease activity in HLH and MAS.24,32 These findings support the therapeutic strategies of targeting pathologically activated IFN-γ–producing T cells (eg, etoposide35 and alemtuzumab36 ) and/or blockade of IFN-γ signaling (eg, IFN-γ antibody emapalumab32 and JAK inhibition [NCT04551131]) for patients with HLH.
To our knowledge, Sumegi et al37,38 have published the only other studies of HLH gene expression to date. They reported increased expression of a wide range of genes involved in immune responses compared with healthy controls and then identified unique clusters of gene expression signatures related to different forms of familial HLH. These studies analyzed RNA from unfractionated peripheral blood mononuclear cells, which may reflect RNA contributions based on relative abundance of particular cell types and differences in cell-specific transcriptional activity. The cell-specific gene expression reported in this study was designed to validate the IFN-γ signature we observed in plasma and identify relative expression of IFN-γ–related genes by cytotoxic T cells and responses of activated monocytes. Extended cell type–specific gene expression studies (and/or single cell sequencing) may provide additional biomarkers for diagnosis, risk stratification, and therapy.
A growing body of research in children with severe sepsis and sepsis-induced multiple organ dysfunction syndrome describes complex immune responses to sepsis, with studies identifying phenotypes of immunoparalysis (with decreased IFN-γ responses), thrombocytopenia-associated multiple organ failure, and MAS associated with death.39-42 Functional immune analyses and biomarker studies among children with severe sepsis are being developed to define pathogenic mechanisms and create clinical models of risk prediction.43,44
The plasma proteome provides a solution to overcome limitations of consensus-driven clinical diagnostic criteria and specialty/physician bias that may confound care for children with hyperinflammatory syndromes with overlapping symptoms. Results from this study support prospective evaluation of diagnostic performance of the 10-protein classifier (CXCL9, CXCL10, CXCL11, IL-1A, IL-6, heparin-binding EGF-like growth factor, CXCL5, migration inhibition factor, and EGF), as well as more focused assays with CXCL9 and IL-6, which are currently commercially available laboratory studies, to differentiate critically ill children with HLH vs severe sepsis/SIRS. Furthermore, this study demonstrates potential for precision approaches to critically ill children with inflammation for diagnosis and therapy. For some patients in the intensive care unit with hyperinflammation, corticosteroids or other immunosuppression may be detrimental.45 However, for others, control of pathologic inflammation may be life saving. This concept is illustrated by reanalysis of data from a phase 3 randomized study of IL-1α blockade with anakinra in patients with severe sepsis. Analyzed as a group, anakinra did not confer any benefit. However, in patients with features of macrophage activation syndrome (hepatobiliary dysfunction/disseminated intravascular coagulation), there was a significant survival advantage.46 Similarly, cytokine profiling predicts risk of pathologic inflammation with cancer immunotherapy, and tocilizumab (targeting IL-6 signaling) shows benefit in patients who develop cytokine release syndrome.47,48 More recently, increased inflammation in patients infected with SARS-CoV-2 (elevated ferritin, elevated IL-6, elevated D-dimer) has been associated with risk of death.49 Moving forward, comprehensive, coordinated multicenter studies with prospective collection and analysis of blood cells and plasma from children who present to the intensive care unit with fever and organ dysfunction may further identify biological and clinical categories between and among the pediatric hyperinflammatory syndromes to inform optimal therapeutic approaches for these challenging patients who are at high risk of death and for whom prompt immunosuppression may be lifesaving (or harmful).
Acknowledgments
The Texas Children's Cancer and Hematology Centers Histiocytosis Program is supported by a research grant from the HistioCure Foundation. This project was further supported by a gift from the Helfman family. Additional relevant grant support includes National Institutes of Health, Specialized Program of Research Excellence in Lymphoma (grant P50CA126752) (to. C.E.A.) and the St. Baldrick’s innovation award (to C.E.A.). This manuscript is a collaborative project with support from St. Baldrick’s Foundation, which sponsors the North American Consortium for Histiocytosis Research (C.E.A., K.M., O.E.E., M.B.J.), Liam’s Lighthouse Foundation (M.B.J., C.E.A., K.M.), the Neuman Family Foundation (M.B.J.), and grant R34HL107801 (M.B.J.).
Authorship
Contribution: H.L., B.P.S., B.R.G., H.A.A., O.E.E., D.J.Z., and T.-K.M. contributed to study design and performed experiments or data analyses and wrote, reviewed, and edited the manuscript; J.L., L.F., I.C., R.C., N.E.M., N.G., J.A., J.G., D.B., K.M., E.M., F.L., T.N., H.W., M.M.H., J.N.G., S.L., M.L.H., L.K.M., M.J., A.N., and M.B.J. contributed clinical data and/or tissue samples and wrote, reviewed, and edited the manuscript; and C.E.A. and M.B.J. conceived of the study, supervised the project, and wrote, reviewed, and edited the manuscript.
Conflict-of-interest disclosure: M.B.J., M.L.H., and C.E.A. have served as consultants for Sobi. M.L.H. serves on a Data Safety Monitoring Committee for Novimmune. All remain authors declare no competing financial interests.
Correspondence: Carl Allen, Baylor College of Medicine, Feigin Center, Suite 730.06, 1102 Bates St, Houston, TX 77030; e-mail: ceallen@txch.org.

![Unsupervised analysis does not distinguish HLH from sepsis/SIRS compared with pediatric controls. (A) Heat map displaying unsupervised clustering of all plasma analytes assayed from noninflammatory pediatric controls, HLH, sepsis, and SIRS. Colors representing specific diagnostic groups/subgroups are included in the legend. Cases of secondary HLH (systemic juvenile idiopathic arthritis [JIA], drug reaction with eosinophilia syndrome [DRESS], and cancer) are indicated. (B) These datasets are also presented using principal component analysis. The control cases clearly separate, whereas HLH, severe sepsis, and SIRS have overlapping inflammatory plasma profiles.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/17/10.1182_bloodadvances.2021004287/2/m_advancesadv2021004287f1.png?Expires=1766469937&Signature=U3H7SWFVOslterEHR2WlkaqbjRTLGiJT9PrYR150Jw7YZfUnDnEwVlE45Ph-j6KRK1aP7pVDgQMKJkGywC~jSOhzYtBVlTVTlH9u29AZNi5xMjWwxAogwcMDyaZTYQqXaR~CV6EJcJUR~YJczH3rD9aQlloBW9BIGcPeMX-ObprdUYv8PGpz7o6okdvlv1cmsYYevzsfQUwiZRJFUxHtODvayXe8ytxx250arGKcbOXag4pkwFbKBaQgO0D5Q-vsuOdfljnO94dZjylfoBkN6-pD302HVVHViem2ZP8mOQV2YvW8~ORdt48fOhU4mfxXMYHm71KZpzRyih3DMF0SQQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)