Key Points
We report clinical evidence of effectiveness using anti-BCMA therapies sequentially.
Careful monitoring of the mechanism of relapse can help to guide clinical decisions about anti-BCMA retreatment.
Abstract
The recent emergence of anti–B-cell maturation antigen (BCMA) therapies holds great promise in multiple myeloma (MM). These include chimeric antigen receptor (CAR) T cells, bispecific antibodies, and antibody-drug conjugates. Their development in clinical trials and further approval are changing the strategy for treating MM. Considering that a cure has not been reached, a central question in the coming years will be the possibility of using these therapies sequentially. Here, we report 2 cases of the serial use of anti-BCMA therapies with parallel monitoring of BCMA expression and anti-CAR antibodies. We further discuss recent data from clinical studies that have informed us about the different mechanisms of resistance to anti-BCMA therapies, including antigen escape, BCMA shedding, anti-drug antibodies, T-cell exhaustion, and the emergence of an immunosuppressive microenvironment. This knowledge will be essential to help guide the strategy of serial treatments with anti-BCMA therapies.
Introduction
The B-cell maturation antigen (BCMA) has emerged as a central target in multiple myeloma (MM). BCMA, also designated as TNFRSF17, belongs to the tumor necrosis factor receptor superfamily, which is a family of cytokine receptors. BCMA’s main ligands are the cytokines B-cell activating factor and a proliferation-inducing ligand.1 The interaction between BCMA and its ligands activates the NF-κB signaling pathway that plays an important role in B-cell proliferation and maturation2 and is essential for the survival of long-lived bone marrow plasma cells.3,4 BCMA is expressed preferentially on mature B cells and has minimal expression on hematopoietic stem cells or other cell types.
In recent years, 3 BCMA-targeting therapies have emerged: bispecific antibodies (BsAbs; (AMG420,5 CC93269,6 and teclistamab7), an antibody-drug conjugate (ADC; belantamab mafodotin8), and chimeric antigen receptor (CAR) T cells (primarily bb2121,9 bb21217,10 JNJ452811 and orvacabtagene autoleucel12). These new anti-BCMA immunotherapies have demonstrated a high efficacy in the context of relapsed/refractory MM. BCMA-targeting CAR T cells are being evaluated in phase 3 trials in MM. The reported phase 1-2 data from the KarMMa,13 CARTITUDE-1,11 EVOLVE,12 and BB2121710 studies reveal overall response rates ranging from 73% to 100% in heavily pretreated patients who received a median of 5 to 6 prior lines of treatment. Of note, the complete remission (CR) rates range from 33% to 86%, with rates of negative minimal residual disease (at 10−5) ranging from 28% to 50%. However, relapses are systematically observed after BCMA-targeting CAR T-cell therapy, even in patients who achieved minimal residual disease negativity, with a median progression-free survival (PFS) of 12.1 months at the target dose of 450 million CAR T cells in the KarMMa14 trial and with a PFS rate of 86% at 9 months in the CARTITUDE-1 study.11 Similar results are seen in dose-escalation studies of BsAbs targeting BCMA and CD3, with ORRs of 67% to 89% and CR rates of 38% to 50% at effective doses.5–7 Response rates are lower with belantamab mafodotin, an anti-BCMA antibody (Ab) conjugated to monomethyl auristatin F, with an ORR of 32% and a PFS of 2.8 months (with 2.5 mg/kg) in the DREAMM-2 phase 2 trial.8 Despite the remarkable efficacy of anti-BCMA immunotherapies, relapses occur systematically, even in patients who achieved deep responses. This observation raises 2 essential questions: What are the mechanisms of resistance to anti-BCMA therapies? and Can we use BCMA-targeting therapies sequentially?
Here, we describe 2 clinical cases of patients receiving sequential anti-BCMA therapies, illustrating the feasibility of this approach. We also discuss the biological and clinical evidence highlighting the mechanism of resistance to anti-BCMA therapies.
Case descriptions
The first case is a 74-year-old man with κ light chain MM, with unknown cytogenetic status, who was treated on a phase 2 trial of BCMA-directed CAR T cells (bb2121, KarMMa13). At enrollment, the patient was refractory to bortezomib, lenalidomide, pomalidomide, and daratumumab, with 4 prior lines of treatment including 2 autograft transplantations. He received lymphodepleting therapy (fludarabine and cyclophosphamide) and was infused with 450 × 106 CAR+ T cells. He achieved a stringent CR at day 30 (Figure 1A) and remained disease free for 1 year before progressing with increased κ light chains, Bence Jones proteinuria, and new bone lesions. CAR T-cell peak expansion exceeded the median observed across treated patients after infusion and was detectable out to 9 months postinfusion. However, CAR T cells dropped below the level of detection in the blood at the time of relapse. The expression of soluble BCMA (sBCMA) declined in parallel with the initial response, but then increased at relapse, suggesting the persistence of BCMA expression by tumor cells. Persistent BCMA expression in >50% of bone marrow plasma cells was confirmed at progression by immunohistochemistry. As allowed per protocol, the patient received a second infusion of 450 × 106 CAR T cells. The monitoring of CAR T cells demonstrated an ∼26-fold lower expansion in response to the second infusion relative to the first infusion (Figure 1A). The patient’s MM did not respond, and the disease continued to progress. A retrospective analysis revealed the presence of anti-CAR antibodies at the time of relapse after the first infusion, potentially contributing to the lower expansion of CAR T cells after the second infusion (Figure 1B). He received subsequent treatment with belantamab mafodotin, a conjugated anti-BCMA Ab (GlaxoSmithKline; compassionate use). The patient achieved a very good partial response after 3 injections of belantamab mafodotin and is still in response 5 months later. In this clinical case, the patient received 3 sequential anti-BCMA therapies: 2 BCMA-targeting CAR T-cell infusions and 1 line of BCMA-targeted ADC. A potential mechanism of failure in response to the second infusion of CAR T cells was the presence of anti-CAR antibodies. The increase in sBCMA and persistent tumor BCMA expression at the time of CAR T-cell failure permitted the effective use of another anti-BCMA therapy with a different single chain variable fragment (scFv). This case illustrates that a systematic evaluation of the mechanism of resistance to anti-BCMA therapies can guide their sequential use in clinical practice.

Treatment course of patients receiving serial BCMA-targeted therapies. CAR T-cell expression and serum free light chain (SFLC) expression (A) and sBCMA and anti-CAR antibodies (B) in patient #1 receiving 2 infusions of BCMA-targeted CAR T cells and belantamab. CAR T-cell expression and circulating plasma cells (C) and sBCMA and anti-CAR antibodies (D) in patient #2 receiving 1 infusion of BCMA-targeted CAR T cells followed by belantamab.
Treatment course of patients receiving serial BCMA-targeted therapies. CAR T-cell expression and serum free light chain (SFLC) expression (A) and sBCMA and anti-CAR antibodies (B) in patient #1 receiving 2 infusions of BCMA-targeted CAR T cells and belantamab. CAR T-cell expression and circulating plasma cells (C) and sBCMA and anti-CAR antibodies (D) in patient #2 receiving 1 infusion of BCMA-targeted CAR T cells followed by belantamab.
The second case is a 68-year-old man with IgG κ MM, with t(14;16), deletion of 17p, and gain of 1q, who was treated on the phase 2 KarMMa trial.13 At enrollment, he had received 4 prior lines of treatment, including 1 autograft transplantation and was refractory to bortezomib, lenalidomide, pomalidomide, and daratumumab. He received bridging therapy with melphalan (50 mg/m2), lymphodepleting therapy (fludarabine and cyclophosphamide), and was infused with 450 × 106 CAR+ T cells. The patient achieved a stringent CR at day 30 and remained disease free for 10 months before progressing with a plasma cell leukemia (Figure 1C). Although bone marrow evaluation at relapse was nonevaluable for tumor BCMA expression, his sBCMA level was high at relapse, reflecting the expression of BCMA by the tumor cells. He also had detectable anti-CAR antibodies at the time of relapse (Figure 1D). He received a seventh line of treatment (carfilzomib, cyclophosphamide, and dexamethasone) with no response and was subsequently treated with belantamab mafodotin (GlaxoSmithKline; compassionate use). He had a rapid response with regard to the peripheral tumor, with a rapid clearance of circulating plasma cells together with a mild tumor lysis syndrome, but he had no response on the M-spike after 2 injections and progressed thereafter (Figure 1C).
These cases emphasize the possibility of using sequential anti-BCMA therapies in BCMA+ relapses, which remain a functional target even after prior therapeutic pressure on this axis. A switch in the antigen binding fragment might be necessary in the case of anti-scFv antibodies. Therefore, careful monitoring of these 2 parameters will be essential to guide treatment decisions in clinical practice in the near future.
Methods
The clinical trial referenced in this article is the KarMMa study,13 which is registered at www.clinicaltrials.gov as #NTC03361748. It was approved by the institutional review board and scientific committee of Centre Hospitalo-Universitaire (CHU) Lille and conducted in accordance with the Declaration of Helsinki.
CAR T-cell expansion/pharmacokinetics
DNA was purified from peripheral blood CD3+ cells. Vector transgene copies were measured per micrograms of genomic DNA by quantitative polymerase chain reaction (qPCR), as previously described.13 CAR T-cell levels were also assessed by an in-house vector-based quantitative polymerase chain reaction (qPcR) assay, as previously described.15
sBCMA evaluation
Baseline and post-CAR T-cell infusion levels of sBCMA were evaluated in patient serum using a Luminex immunoassay (cat. no. LXSAHM-01; R&D Systems).
Anti-drug antibodies
The potential immune response to idecabtagene vicleucel was evaluated for humoral responses. Serum samples collected postinfusion were evaluated for the formation of anti-drug antibodies using an immunoassay that was designed and validated to detect antibodies to the extracellular CAR domain, as previously described.13
Results and discussion
The recent development of multiple anti-BCMA immunotherapies has raised the question of using them sequentially. Emerging clinical experience supports the effectiveness of this approach. In addition to the 2 cases reported here, 5 cases were described previously in the literature.16,17 Among them, 3 received murine-scFv anti-BCMA CAR T cells, followed by fully human BCMA CAR T-cell therapy; 2 patients achieved a CR, and 1 achieved a very good partial response.16
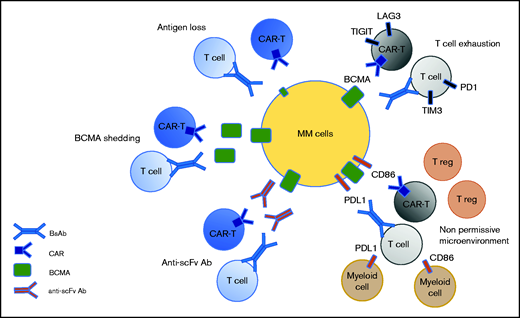
Recent data from clinical studies have informed us about the different mechanisms of resistance to anti-BCMA therapies. Some are antigen dependent, such as antigen escape, BCMA shedding, and anti-scFv antibodies. Others are T-cell dependent, such as T-cell exhaustion and the emergence of an immunosuppressive microenvironment (Figure 2). Antigen escape has been reported but it does not appear to be frequent in the context of anti-BCMA therapies, probably because it is essential for the survival of plasma cells.3 In the KarMMa study, of 16 patients with an evaluable bone marrow sample at relapse, only 1 patient had an antigen loss.18 Biallelic deletion on chromosome 16, encompassing the BCMA locus, has been described as a mechanism of BCMA loss.19,20 BCMA can also undergo γ-secretase–mediated shedding from plasma cells, leading to the circulation of sBCMA.21 Theoretically, high levels of sBCMA could interfere with anti-BCMA therapies by coating a BCMA-directed binding fragment (scFv or Ab) and, thereby, function as an antigen-masking mechanism.22 However, no clinical evidence exists to suggest that sBCMA levels can negatively impact BCMA-targeting therapies, but additional clinical experience, including testing γ-secretase inhibitors, will help to elucidate the effect of sBCMA levels on the function of BCMA-targeting therapies. Until recently, most of the BCMA-targeting CAR T cells evaluated in the clinic have derived their scFv from nonhuman species (mouse for bb21219 and camelid for JNJ452811). The use of nonhuman scFv can induce immunogenicity resulting from an adaptive immune response after CAR T-cell infusion, which may play a role in limiting the persistence of the CARs. A recent study of 17 patients with relapsed/refractory MM treated with the biepitopic BCMA-targeting CAR T-cell therapy ciltacabtagene autoleucel revealed that 7 of them had developed high levels of anti-CAR antibodies.23 The incidence of relapse was significantly higher than that in patients without detectable humoral immunogenicity. Therefore, anti-CAR antibodies are associated with a high risk for relapse after CAR T-cell therapy. The presence of anti-scFv antibodies in patients relapsing following BCMA-targeting CAR T-cell therapy also emphasizes the strategy of developing fully human or humanized scFv and antibodies.
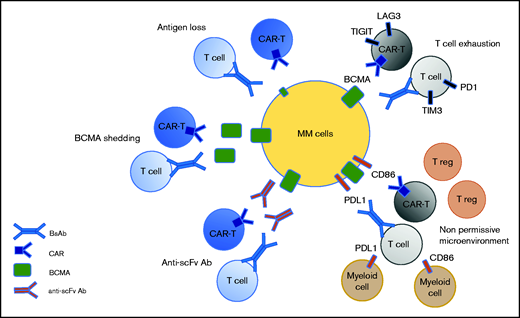
Potential mechanism of resistance to anti-BCMA immunotherapies. Three mechanisms are target dependent, including antigen loss, BCMA shedding by sBCMA, and the secretion of anti-drug antibodies. Two mechanisms of resistance are T-cell dependent, including T-cell exhaustion and the emergence of a nonpermissive microenvironment.
Potential mechanism of resistance to anti-BCMA immunotherapies. Three mechanisms are target dependent, including antigen loss, BCMA shedding by sBCMA, and the secretion of anti-drug antibodies. Two mechanisms of resistance are T-cell dependent, including T-cell exhaustion and the emergence of a nonpermissive microenvironment.
T-cell exhaustion remains a potential major mechanism of relapse during BCMA-targeting therapies. In the context of CAR T cells or BsAbs, responses have been associated with a higher CD4/CD8 ratio and an increased frequency of CD45RO−CD27+CD8+ T cells, reflective of stem memory T cells.24,25 In contrast, T cells with exhausted or senescent phenotypes are enriched in patients who are resistant to anti-BCMA BsAb or CAR T cells.25 This indicates that the efficacy of BCMA-targeting therapies may be greater during early stages of the disease when patients are less immunosuppressed. Moreover, little is known regarding the response of the tumor microenvironment to immunotherapies and its capacity to impact the response to a subsequent immunotherapy. Further studies with sequential sampling will help to determine the role of the microenvironment in anti-BCMA failure.
Knowledge about the mechanisms of resistance to anti-BCMA therapies will help to define how we can use them sequentially. BCMA+ relapses represent an opportunity for sequential treatment using anti-BCMA therapies. The switch in antigen binding fragment prevents the inhibition of subsequent anti-BCMA therapies by anti-drug antibodies developed during a previous exposure. Another aspect, but less well explored, is the impact of T-cell failure, as well as an immunosuppressive microenvironment, and how it can affect retreatment with anti-BCMA CAR T-cell or BsAb therapies. Monitoring of exploratory biomarkers, such as sBCMA, tumor BCMA expression, and the emergence of anti-drug antibodies, will help to further define the major mechanisms of resistance and guide clinicians in making therapeutic decisions.
Authorship
Contribution: All authors contributed to the outline, design, writing, and editing of the manuscript.
Conflict-of-interest disclosure: I.Y.-A. has received honorarium from Celgene/Bristol Myers Squibb, Janssen, Kite/Gilead, and Novartis. T.B.C. works for Bristol Myers Squibb corporation. T.F. is a member of the speaker’s bureau for Celgene/Bristol Myers Squibb and Janssen. S. Manier has received research funding from AbbVie, Amgen, Celgene/Bristol Myers Squibb, GlaxoSmithKline, Janssen, and Takeda. The remaining authors declare no competing financial interests.
Correspondence: Salomon Manier, Service d’Hématologie, Hôpital Huriez, CHU Lille, rue Michel Polonovski, 59000 Lille, France; e-mail: salomon.manier@chru-lille.fr.