Key Points
Targeted sequencing of ccfDNA can detect clinically relevant mutations missed by conventional bone marrow analysis.
ccfDNA and bone marrow sequencing may play complementary roles in patients with AML.

Abstract
Circulating cell-free DNA (ccfDNA) allows for noninvasive peripheral blood sampling of cancer-associated mutations and has established clinical utility in several solid tumors. We performed targeted next-generation sequencing of ccfDNA and bone marrow at the time of diagnosis and after achieving remission in 22 patients with acute myeloid leukemia (AML). Among 28 genes sequenced by both platforms, a total of 39 unique somatic mutations were detected. Five mutations (13%) were detected only in ccfDNA, and 15 (38%) were detected only in bone marrow. Among the 19 mutations detected in both sources, the concordance of variant allelic frequency (VAF) assessment by both methods was high (R2 = 0.849). Mutations detected in only 1 source generally had lower VAF than those detected in both sources, suggesting that either method may miss small subclonal populations. In 3 patients, sequencing of ccfDNA detected new or persistent leukemia-associated mutations during remission that appeared to herald overt relapse. Overall, this study demonstrates that sequencing of ccfDNA in patients with AML can identify clinically relevant mutations not detected in the bone marrow and may play a role in the assessment of measurable residual disease. However, mutations were missed by both ccfDNA and bone marrow analyses, particularly when the VAF was <10%, suggesting that ccfDNA and bone marrow may be complementary in the assessment and monitoring of patients with AML.
Introduction
Accurate molecular profiling in acute myeloid leukemia (AML) and other leukemias is vital for accurate risk stratification and selection of targeted therapies.1 Sequential monitoring of somatic mutations in remission samples also aids in the assessment of measurable residual disease (MRD). After initial therapy, the presence of persistently detectable leukemia-associated mutations or other genomic alterations as assessed by either polymerase chain reaction (PCR) or next-generation sequencing (NGS) is associated with higher rates of relapse and worse survival in patients with AML.2-6 Although the optimal intervention for patients who have persistent or recurrent MRD is still largely unknown, allogeneic stem cell transplantation (alloSCT), MRD-directed clinical trial, or other change in therapy should be considered for these patients, as the risk of relapse in the absence of any intervention is very high.7 Assays that can reliably detect residual mutations in patients in morphological and hematological remission are therefore imperative in the management of AML.
MRD monitoring is most commonly performed on bone marrow aspirates, which requires an invasive procedure. Although some studies have evaluated peripheral blood monitoring of MRD, these have been largely restricted to the evaluation of specific targets using PCR (eg, NPM1 mutations).3,8,9 Circulating cell-free DNA (ccfDNA) is highly fragmented DNA in plasma that is released by normal or tumor cells that undergo apoptosis or necrosis and allows for noninvasive peripheral blood sampling of cancer-associated mutations.10 Detection of mutations in ccfDNA is informative in various solid tumors, including as a marker of MRD,11,12 but its role in AML and other leukemias is largely unexplored. In 1 study of ccfDNA sequencing in patients with AML or myelodysplastic syndrome (MDS), persistence of leukemia-associated mutations after alloHSCT was associated with significantly higher cumulative incidence of relapse.13 Despite these promising results, it is unknown whether ccfDNA can fully supplant bone marrow assessment for molecular profiling and MRD measurement in AML. In this study, we therefore sought to assess the relative utility of baseline assessment and tracking of leukemia-associated mutations through peripheral blood sampling of ccfDNA, as compared with standard assessment on bone marrow specimens, in patients with AML.
Methods
Study design and participants
This was a prospective study evaluating the utility of ccfDNA and bone marrow sources for molecular profiling in patients with AML. This study was conducted at a single academic center (The University of Texas MD Anderson Cancer Center). Patients with newly diagnosed AML (excluding acute promyelocytic leukemia) who were undergoing frontline intensive chemotherapy (defined as a cumulative dose of cytarabine ≥4 g/m2 in induction) were eligible. Baseline bone marrow aspiration was performed prior to initiating chemotherapy, with sequential bone marrow assessments performed periodically as standard of care. Peripheral blood for ccfDNA analysis was performed at baseline and during remission in a subset of patients. All ccfDNA testing was performed blinded to the results from bone marrow analysis. This study was approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center. All patients provided informed consent according to institutional guidelines and the Declaration of Helsinki.
Bone marrow sample processing and analysis
Mutation analysis was performed on bone marrow specimens containing morphological disease (ie, >5% blasts) using a 28-gene targeted NGS panel in our clinical laboratory as previously described.14,15 Genomic DNA was extracted from bone marrow aspirates. Amplicon-based NGS targeting the entire coding regions of a panel of 28 genes associated with myeloid neoplasms was performed using a MiSeq platform (Illumina, San Diego, CA). The genes analyzed included the following: ABL1, ASXL1, BRAF, DNMT3A, EGFR, EZH2, FLT3, GATA1, GATA2, HRAS, IDH1, IDH2, IKZF2, JAK2, KIT, KRAS, MDM2, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PTPN11, RUNX1, TET2, TP53, and WT1. All coding exons for each gene are covered (supplemental Table 1). A minimum sequencing coverage of 250× (bidirectional true paired-end sequencing) and minimum input of 250 ng of DNA were required. The analytical sensitivity was established at 5% mutant reads in a background of wild-type reads. Established bioinformatics pipelines were used to identify somatic variants.
ccfDNA sample processing and analysis
Peripheral blood was collected in Streck tubes. Ten milliliters of blood was collected at each time point, and at least 20 ng of extracted DNA was required for sequencing. Following plasma isolation by centrifugation, ccfDNA was extracted by automated methods that employ the silica-based magnetic beads and with the use of the QIAamp circulating nucleic acid kit (Qiagen, Germantown, MD). Targeted NGS was performed using a 275-gene panel (supplemental Table 2). Among the 28 genes that overlap with the bone marrow sequencing assay, the coverage included all coding exons as shown in supplemental Table 1. The panel is based on single primer extension library preparation (Qiagen). Genomic DNA samples are first fragmented, end repaired, and A-tailed within a single, controlled multienzyme reaction. Prior to target enrichment and library amplification, each original DNA molecule is assigned a unique sequence or index, or unique molecular identifier (UMI). This assignment is accomplished by ligating fragmented DNA with an adapter containing a 12-base fully random sequence (ie, the UMI). Target enrichment is performed post-UMI assignment to ensure that DNA molecules containing UMIs are sufficiently enriched in the sequenced library. For enrichment, ligated DNA molecules are subjected to several cycles of targeted PCR using 1 region-specific primer and 1 universal primer complementary to the adapter single primer extension. A universal PCR is ultimately carried out to amplify the library and add platform-specific adapter sequences and additional sample indices. The sequencing is conducted using the Illumina NextSeq 550 instrument and associated reagents. Reads are aligned in paired end mode to the GRCh37 p13 version of the human genome. Aligned reads are written to a Binary Alignment Map format from the DRAGEN aligner. PCR duplicates are marked. The DRAGEN somatic analysis pipeline identifies 2 classes of mutations, as follows: (1) single nucleotide variants and (2) indels. Mixing studies and comparative studies with paired bone marrow samples were performed to establish reproducibility of the ccfDNA assay (supplemental Figure 1; supplemental Table 3). The overall mean coverage is >1000×, and a sequence coverage ≥100× (after removing duplicates) is required for a specific mutation to be called. The analytical sensitivity of the 275-gene panel was established at 5% allelic frequency for non–hot spots and at 3% allele frequency for hot spots. Hot spots were defined as mutations in KRAS (12,13,61), ASXL1 (Glu391), NRAS (12,13,61), FLT3 (Glu598), ABL1 (T315), and JAK2 (V617F).
MRD analysis
In remission samples, ccfDNA sequencing data were compared with results from clinical MRD assays performed at the same time point. Flow MRD was assessed by 8-color multiparameter flow cytometry as previously described.16 Fluorescence in situ hybridization and PCR for relevant targets as well as chimerism studies were performed as previous described.17,18
Statistical methods
Patient characteristics were summarized using median (range) for continuous variables and frequencies (percentages) for categorical variables. Associations between categorical and continuous variables were assessed using χ2 tests and 1-way analysis of variance. Concordance of bone marrow and ccfDNA results were assessed using Pearson correlation calculation. For analysis, only the 28 genes sequenced by both the bone marrow and the ccfDNA targeted sequencing panels were used. All statistical tests were performed using GraphPad Prism 6.
Results
Patient characteristics and sample collection
Twenty-two patients with newly diagnosed AML were evaluated. Baseline characteristics of the study cohort are shown in Table 1. Three patients had a history of prior malignancy (breast cancer, soft tissue sarcoma, and diffuse large B-cell lymphoma in 1 patient each), all of whom had no evidence of disease at the time of AML diagnosis. Eight patients (36%) had absolute peripheral blood blast count <1 × 109/L, and 1 patient had no peripheral blood circulating blasts. All 22 patients had paired ccfDNA and bone marrow targeted sequencing performed at baseline. For baseline samples, ccfDNA was collected a median of 6 days after bone marrow collection (range, 0 to 17 days), and a median of 1 day after start of induction chemotherapy (range, −7 to 7 days). Nine patients (41%) also had ccfDNA collected at ≥1 time point during remission.
Concordance of mutation detection in ccfDNA and bone marrow in baseline samples
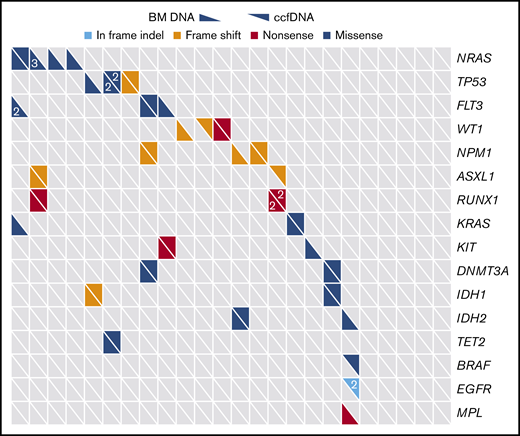
Among the 28 genes sequenced by both assays, a total of 39 unique somatic mutations were detected (Figure 1). Retrospective manual review of raw sequencing data was performed in all discordant cases in order to rule out possible discrepancies due to slight differences in the bioinformatics pipelines of the 2 sequencing methods. Thirty-four of these mutations were detected in the bone marrow, and 24 were detected in ccfDNA (supplemental Table 4). Nineteen mutations (49%) were detected by both methods; 5 (13%) were detected only in ccfDNA, and 15 (38%) were detected only in the bone marrow. In the 3 patients in whom ≥1 mutation was detected in ccfDNA but not in the bone marrow, all had circulating blasts at the time of ccfDNA collection; however, the peripheral blood absolute blast count of these patients (0.03, 0.11, and 2.79 × 109/L, respectively) was lower than the median for the entire cohort, suggesting that differences in quantities of circulating blasts were unlikely to account for this discrepancy. Among the 28 overlapping genes, the median number of mutations per patient detected in bone marrow and in ccfDNA was 1 for both assays (range, 0 to 5 and 0 to 3, respectively; P = .30). In comparison with patients in whom ccfDNA was collected prior to the start of chemotherapy and those in whom ccfDNA was collected after the start of chemotherapy, there was no difference in the concordance/discordance rate of mutation detection between the 2 assays (P = .72), suggesting that time between ccfDNA collection and treatment initiation was not a confounder in this analysis.
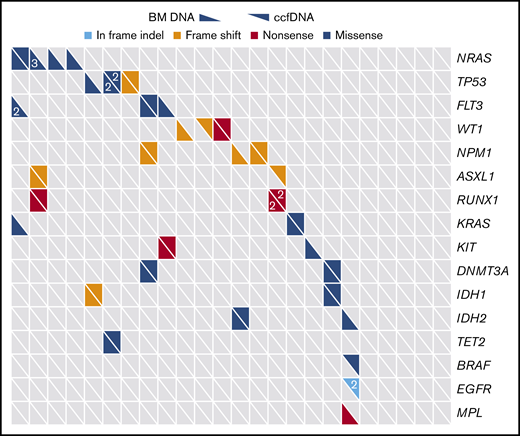
Distribution of mutations as detected by targeted sequencing of ccfDNA and bone marrow (BM). Only genes sequenced by both assays are included. Each column represents a unique patient with AML (N = 22), and each row represents a gene that was detected in at least 1 patient. Numbers, when present, indicate when >1 mutation of a given gene were detected in the same patient.
Distribution of mutations as detected by targeted sequencing of ccfDNA and bone marrow (BM). Only genes sequenced by both assays are included. Each column represents a unique patient with AML (N = 22), and each row represents a gene that was detected in at least 1 patient. Numbers, when present, indicate when >1 mutation of a given gene were detected in the same patient.
Impact of variant allelic frequency (VAF) on mutation discordance in baseline samples
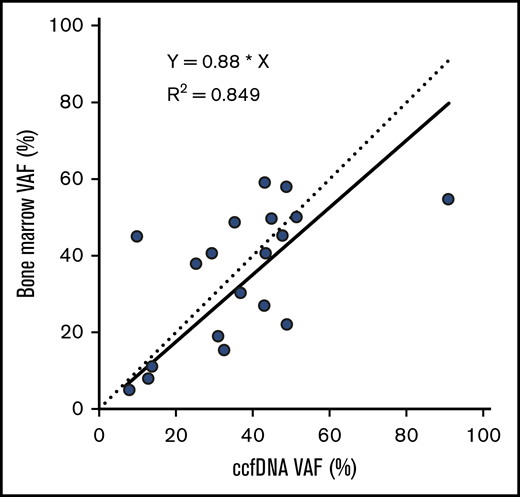
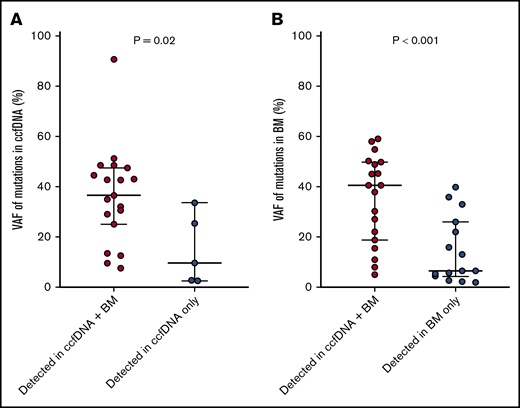
The median VAF of mutations detected in ccfDNA and in bone marrow was 33.0% (range, 2.7% to 90.8%) and 24.1% (range, 2.0% to 59.2%), respectively. Among the 19 individual mutations detected by both assays, the concordance of VAF assessment by both methods was high (R2 = 0.849; Figure 2). Mutations detected by only one of the 2 methods were generally of lower VAF than those detected by both methods, suggesting that either method may miss small subclonal populations. The median VAF of mutations (as measured in ccfDNA) that were detected by both methods was higher than those detected only in ccfDNA (36.7% vs 9.8%, respectively; P = .02; Figure 3A); similarly, the median VAF of mutations (as measured in the bone marrow) that were detected by both methods was higher than those detected only in the bone marrow (40.7% vs 6.6%; P < .001; Figure 3B). The VAF was <10% in 3 of the 5 mutations (60%) detected in ccfDNA but not in bone marrow and in 8 of the 15 mutations (53%) detected in bone marrow but not in ccfDNA.
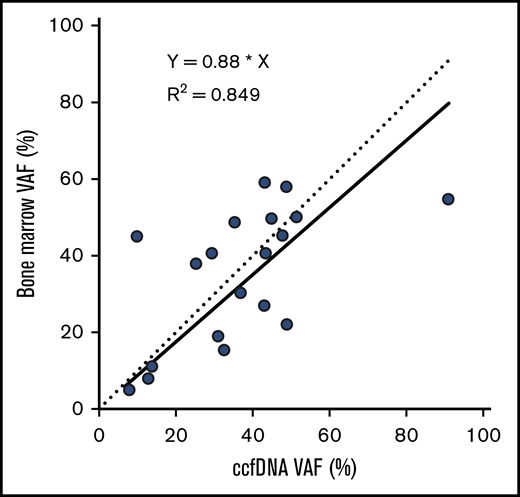
VAFs detected in ccfDNA and in bone marrow, among the 19 mutations detected by both assays. The solid line represents the correlation of the linear regression fit of the data, and the dotted line represents a 1:1 ratio for reference.
VAFs detected in ccfDNA and in bone marrow, among the 19 mutations detected by both assays. The solid line represents the correlation of the linear regression fit of the data, and the dotted line represents a 1:1 ratio for reference.
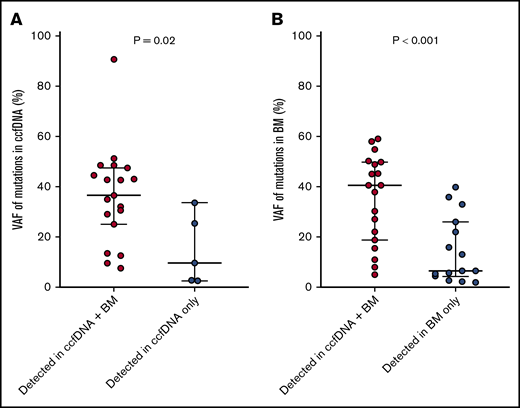
VAFs of mutations detected in both ccfDNA and in bone marrow vs those detected in only 1 source. VAFs of mutations detected in both ccfDNA and in bone marrow vs those detected only in ccfDNA (A), and detected in both ccfDNA and in bone marrow vs those detected only in bone marrow (B).
VAFs of mutations detected in both ccfDNA and in bone marrow vs those detected in only 1 source. VAFs of mutations detected in both ccfDNA and in bone marrow vs those detected only in ccfDNA (A), and detected in both ccfDNA and in bone marrow vs those detected only in bone marrow (B).
Utility of ccfDNA targeted sequencing in baseline and remission samples
Among the 5 baseline mutations detected only by ccfDNA, mutation of ASXL1 was detected in 1 patient (unique patient number [UPN] 1), WT1 in 1 patient (UPN 15), and BRAF and 2 EGFR mutations in 1 patient (UPN 22). Among the 3 patients in whom mutations were detected in ccfDNA but not in bone marrow, none eventually relapsed, and therefore, repeat bone marrow testing in the setting of active disease was not possible. None of these patients is known to have subsequently developed a second malignancy that could account for the discrepant mutations detected in ccfDNA but not the bone marrow. However, notably, the patient with BRAF and EGFR mutations (a 33-year-old man) was lost to follow-up after receiving induction and consolidation and died ∼2 years later (cause of death unknown).
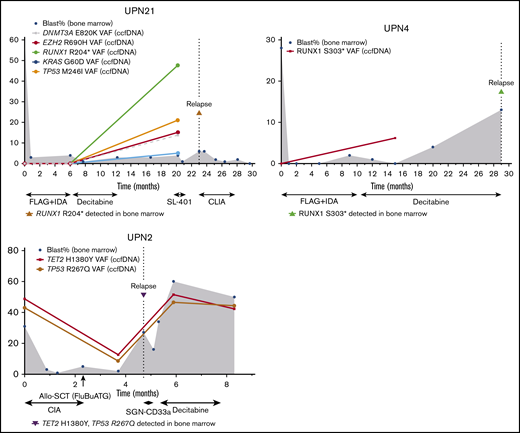
ccfDNA also detected leukemia-associated mutations during remission that appeared to herald overt relapse (Figure 4). Two patients with t(8;21) AML developed new RUNX1 mutations detected by ccfDNA while in remission and subsequently relapsed 3 months (UPN 21) and 14 months later (UPN 4). In both of these patients, the new RUNX1 mutation was not detected in either ccfDNA or bone marrow at baseline and was confirmed in the bone marrow at the time of morphological relapse. In the former patient (UPN 21), remission bone marrow performed concomitantly with ccfDNA collection showed equivocal MRD by multiparameter flow cytometry (0.2% aberrant myeloblasts but partially different immunophenotype from original) and negative fluorescence in situ hybridization for RUNX1-RUNX1T1. PCR for RUNX1-RUNX1T1 was not performed in this case. In the latter patient (UPN 4), concomitant bone marrow was negative for MRD by flow cytometry but showed very low-level RUNX1-RUNX1T1 transcripts by PCR at a level of <0.01%. Another patient with AML had persistent TP53 and TET2 mutations detected by ccfDNA 1 month after alloSCT and subsequently relapsed 1 month later (UPN 2). In this patient, bone marrow and chimerism studies performed at the time of ccfDNA collection showed positive MRD by flow cytometry (0.3%) and microsatellite polymorphism pattern consistent with mixed chimerism (95% of the total DNA, 87% of T cells, and 98% of myeloid cells of donor origin).
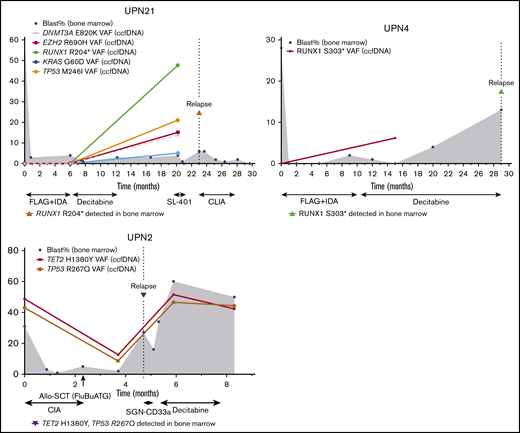
Three cases in which new or persistent mutations detected by ccfDNA predicted subsequent relapse. In cases UPN21 and UPN4, new mutation(s) detected by ccfDNA while in remission preceded overt morphologic relapse. In case UPN2, persistent mutations after allogeneic stem cell transplant were detected 1 month prior to morphologic relapse. Treatment regimens are shown below the x-axis. The y-axis represents percentage of bone marrow blasts and VAFs in the ccfDNA and/or bone marrow. The solid line represents ccfDNA VAFs over time. The dotted vertical line represents the time of relapse. The shaded gray area represents bone marrow blasts over the course of therapy and monitoring. CLIA, cladribine, idarubicin, and cytarabine; FLAG+IDA, fludarabine, cytarabine, granulocyte-stimulating factor, and idarubicin.
Three cases in which new or persistent mutations detected by ccfDNA predicted subsequent relapse. In cases UPN21 and UPN4, new mutation(s) detected by ccfDNA while in remission preceded overt morphologic relapse. In case UPN2, persistent mutations after allogeneic stem cell transplant were detected 1 month prior to morphologic relapse. Treatment regimens are shown below the x-axis. The y-axis represents percentage of bone marrow blasts and VAFs in the ccfDNA and/or bone marrow. The solid line represents ccfDNA VAFs over time. The dotted vertical line represents the time of relapse. The shaded gray area represents bone marrow blasts over the course of therapy and monitoring. CLIA, cladribine, idarubicin, and cytarabine; FLAG+IDA, fludarabine, cytarabine, granulocyte-stimulating factor, and idarubicin.
Discussion
We performed targeted NGS of both bone marrow and ccfDNA in patients with AML and found that NGS of ccfDNA can identify potentially clinically relevant mutations not detected by bone marrow sequencing. Sequential sampling of ccfDNA of patients in complete hematological and morphological remission also identified patients with new or persistent mutations that appeared to signal impending relapse. Together these findings highlight the potential utility of noninvasive ccfDNA sampling at the time of diagnosis and sequentially assessed over the course of therapy.
Accurate assessment of somatic mutations is vital in the management of patients with AML and other leukemias, as this information can guide prognostication and therapeutic decision making, including the recommendation for alloSCT in first remission or the introduction of targeted therapies (eg, for patients with FLT3, IDH1, or IDH2 mutations).1,19 Although it is theoretically attractive to supplant molecular assessment of the bone marrow with less-invasive ccfDNA, these results suggest that such an approach may miss an unacceptable number of true mutations (38% in this cohort). Somatic mutations were missed by both ccfDNA and bone marrow analyses, particularly when the VAF was low, suggesting that bone marrow and ccfDNA sources may be complementary in the assessment and monitoring of patients with AML. Furthermore, as the number of targetable mutations grows in AML and other leukemias, a 10% to 15% false negative rate with standard bone marrow sampling could lead to suboptimal therapy being selected for many patients. It is important to note however that we cannot confirm that all 5 mutations detected in ccfDNA but not in bone marrow were in fact true mutations. Although it is theoretically possible that these discrepant mutations detected in ccfDNA but not in bone marrow could be from other neoplastic tissues, it is notable that only 3 patients had any prior history of solid malignancy, and none had any residual disease at the time of AML diagnosis and ccfDNA collection. Furthermore, even in the context of radiographically measurable solid tumors, the level of detectable mutations by ccfDNA is generally <1%, which is below the level of detection of our ccfDNA assay, decreasing the likelihood that our discrepant findings could be explained by a second occult malignancy.20 Nevertheless, in the 3 patients harboring mutations detected in ccfDNA but not in bone marrow, we cannot definitely rule out that these mutations are not derived from other nonleukemic sources, including (but not limited to) preneoplastic tissues. This possible explanation may be particularly relevant for the patient in whom 1 BRAF and 2 EGFR mutations were detected by ccfDNA but who was lost to follow-up and then subsequently died of an unknown cause, as these mutations are very rare in AML.
A major potential advantage of ccfDNA assessment in AML is as a marker of MRD. Several studies have suggested that NGS-based assessment of the bone marrow for residual leukemia-associated mutations is prognostic for risk of relapse and overall survival.4-6 However, compared with genomic assessment on bone marrow specimens, ccfDNA has the advantage of being collected relatively noninvasively (obtained through peripheral blood) and may therefore be more feasible to assess longitudinally. In some hematological malignancies, ccfDNA assessment has been suggested to be a reliable measure of residual disease,21,22 including in 1 study of AML after alloSCT.13 Although our study was not powered to evaluate the prognostic impact of MRD as detected by ccfDNA sequencing, our description of 3 cases in which emergent or persistent mutations appeared to predict for subsequent relapse provides further support for MRD assessment using ccfDNA.
This study has several limitations. First, we evaluated only 22 patients, and remission samples were obtained at heterogenous time points; therefore, our ability to correlate molecular MRD with risk of relapse and other relevant outcomes is limited. We also used 2 different sequencing platforms for analysis of bone marrow and ccfDNA. Although the ccfDNA panel included 275 different genes, we chose to focus our analysis on the 28 genes that overlapped between these 2 assays, which better allows for assessment of concordance and discordance between these 2 sources. Because of slight differences in analysis between the bone marrow and ccfDNA sources, it is theoretically possible that this could have confounded our results. However, the lowest detected mutation VAF was similar between the 2 sources (2.0% with bone marrow and 2.7% with ccfDNA) and the association of VAF between these 2 methods was good (R2 = 0.849), suggesting similar analytic sensitivity and good concordance. Of particular interest are the cases in which mutations were detected in 1 source but not the other. Without high-sensitivity validation, it is not possible to know with certainty whether these are more accurately classified as false positives or false negatives. High sensitivity NGS or PCR would be needed to confirm that these discordant mutations are truly present and are not in fact sequencing errors. The sensitivity of ccfDNA analysis can be increased significantly if used for monitoring specific mutations; however, in this study, the ccfDNA testing was performed blindly to reduce bias when assessing concordance and discordance of the 2 sequencing methods. In principle, with the use of UMI and by focusing on mutations detected in the diagnostic sample, a sensitivity of 1:1000 can be reliably achieved in detecting mutations by NGS, improving its reliability as a potential MRD assay.23 A prospective comparison with sequencing of peripheral blood mononuclear cells could also be informative. Although this study was not designed to make this direct comparison, a previous study in patients with MDS has suggested that sequencing of ccfDNA is likely superior to sequencing of cellular peripheral blood DNA.24
In conclusion, targeted sequencing of ccfDNA and bone marrow provided complementary genomic information in patients with AML, which may be particularly relevant in the context of subclonal mutations with lower VAF. Sequential sampling of ccfDNA identified new or persistent mutations that appeared to herald overt relapse, suggesting that ccfDNA could be used as a relatively noninvasive source to track leukemia-associated mutations and potentially provide prognostic information about risk of subsequent relapse. These findings provide rationale to evaluate NGS of ccfDNA in larger, prospective cohorts as a marker of MRD in AML and other leukemias.
Data will be shared via e-mail by request of the corresponding author, Farhad Ravandi; e-mail: fravandi@mdanderson.org.
Acknowledgments
This work was supported by an MD Anderson Cancer Center Support Grant (CA016672 from the National Institutes of Health, National Cancer Institute) and SPORE and by the K12 Paul Calabresi Clinical Oncology Scholar Award (N.J.S.) and the American Society of Hematology Junior Faculty Scholar Award in Clinical Research (N.J.S.). Genomic Testing Cooperative Laboratories provided sequencing at no cost.
Authorship
Contribution: N.J.S., K.P.P., and F.R. designed the study, collected and analyzed the data, and wrote the manuscript; K.P.P., M.A., R.L., R.K.-S., J.M., and W.M. collected and sequenced the samples and analyzed the data; M.F., R.A., S.L., and F.W. collected and analyzed the data; G.M.-B., K.T., G.C.I., S.M.K., E.J., G.G.-M., H.M.K., and Z.E. enrolled patients; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: M.A. and W.M. are employees of Genomic Testing Cooperative, which provided sequencing. The remaining authors declare no competing financial interests.
Correspondence: Farhad Ravandi, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Unit 428, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: fravandi@mdanderson.org.