Key Points
GVHD biomarkers can be used as real-time inclusion criteria to selectively target asymptomatic, high-risk patients for intervention.
Preemptive treatment with ΑΑΤ did not improve GVHD outcomes.

Abstract
Steroid-refractory (SR) acute graft-versus-host disease (GVHD) remains a major cause of nonrelapse mortality (NRM) after allogeneic hematopoietic cell transplantation (HCT), but its occurrence is not accurately predicted by pre-HCT clinical risk factors. The Mount Sinai Acute GVHD International Consortium (MAGIC) algorithm probability (MAP) identifies patients who are at high risk for developing SR GVHD as early as 7 days after HCT based on the extent of intestinal crypt damage as measured by the concentrations of 2 serum biomarkers, suppressor of tumorigenesis 2 and regenerating islet-derived 3α. We conducted a multicenter proof-of-concept “preemptive” treatment trial of α-1-antitrypsin (AAT), a serine protease inhibitor with demonstrated activity against GVHD, in patients at high risk for developing SR GVHD. Patients were eligible if they possessed a high-risk MAP on day 7 after HCT or, if initially low risk, became high risk on repeat testing at day 14. Thirty high-risk patients were treated with twice-weekly infusions of AAT for a total of 16 doses, and their outcomes were compared with 90 high-risk near-contemporaneous MAGIC control patients. AAT treatment was well tolerated with few toxicities, but it did not lower the incidence of SR GVHD compared with controls (20% vs 14%, P = .56). We conclude that real-time biomarker-based risk assignment is feasible early after allogeneic HCT but that this dose and schedule of AAT did not change the incidence of SR acute GVHD. This trial was registered at www.clinicaltrials.gov as #NCT03459040.
Introduction
Acute graft-versus-host disease (GVHD) requiring systemic steroid treatment develops in ∼45% of allogeneic hematopoietic cell transplant (HCT) recipients and remains a major cause of nonrelapse mortality (NRM).1,2 GVHD of the gastrointestinal (GI) tract that is resistant to steroid treatment is the primary driver of NRM, but its development cannot be accurately predicted by pretransplant clinical characteristics.3 Steroid-refractory (SR) GVHD is a strong surrogate for long-term outcomes such as NRM and survival that can be calculated within 100 days of HCT.4 Strategies that rely upon intensification of GVHD prophylaxis to prevent the development of SR GVHD, such as the addition of pre-HCT anti-thymocyte globulin (ATG) or prednisone, are insufficiently selective, do not consistently reduce GVHD, and expose patients to additional risks such as increased infections and relapse.5-9 One potentially more attractive approach is to selectively intensify treatment in those patients who are at high risk but have not yet developed GVHD after HCT. However, such a preemptive approach requires both a method that accurately identifies patients who are at high risk for severe GVHD and an intervention that can improve their outcomes.
The Mount Sinai Acute GVHD International Consortium (MAGIC) group of HCT centers has previously validated an algorithm that uses the serum concentrations of 2 GVHD biomarkers measured prior to the onset of GVHD symptoms and as early as day 7 after HCT to predict severe GVHD and NRM more accurately than clinical risk factors such as conditioning intensity, donor type, or HLA match.10 The 2 biomarkers measured in the MAGIC algorithm probability (MAP) are suppressor of tumorigenesis 2 (ST2) and regenerating islet-derived 3α (REG3α). These proteins are released into the circulation by damaged GI crypts and can serve as a liquid biopsy that measures the extent of mucosal damage in the intestine prior to the onset of GVHD symptoms.
The ideal agent for a preemptive treatment strategy, especially one administered early after HCT, would offer protection with minimal toxicity. α-1-Antitrypsin (AAT) is a serine protease inhibitor with anti-apoptotic, anti-inflammatory, and immunomodulatory properties.11-14 It is approved by the US Food and Drug Administration for the treatment of AAT deficiency and has been widely administered, with few side effects. The most common adverse reactions in clinical trials were headaches and upper respiratory infections (<1% of infusions). AAT has shown promise as a potential treatment of GVHD in murine models, where it significantly reduced serum levels of proinflammatory cytokines, suppressed GVHD, and increased survival.15,16 AAT is currently being studied for prevention of GVHD (NCT03805789) and as primary treatment of GVHD (NCT04167514). In addition, 2 phase 2 clinical trials of AAT in patients with SR GVHD demonstrated response rates as high as 65%.17,18
In this multicenter study, we evaluated serum MAPs at day 7 and day 14 post-HCT to identify patients at high risk for developing SR GVHD to determine the feasibility, safety, and efficacy of this novel strategy. Patients were identified within 24 hours as high risk by their MAP on either day 7 or 14 after HCT, and those who did not have any GVHD symptoms were preemptively treated with AAT and prospectively monitored both for safety and for the development of SR GVHD. We then compared the outcomes in patients who received preemptive treatment to a near-contemporaneous control cohort of patients at high risk by MAP from the MAGIC database/biorepository.
Methods
This limited institution study was approved by the institutional review board at each of the 5 participating centers and was coordinated by the MAGIC Data Coordinating Center of the Icahn School of Medicine at Mount Sinai. Informed consent was obtained from all subjects. This trial was registered at www.clinicaltrials.gov as #NCT03459040. Subjects were recruited and enrolled between August 2018 and July 2019.
Subjects
Subjects were recruited from MAGIC centers where the procedures for obtaining screening samples for biomarker scoring has already been established (supplemental Table 1). Eligible subjects were high risk by MAP at day 7 or 14 after HCT and were between the ages of 18 and 75 years. All patients had received allogeneic HCT from related or unrelated donors with at least a 7/8 HLA match. Inclusion criteria permitted any conditioning regimen, stem cell source (peripheral blood or bone marrow), and GVHD prophylaxis, including the use of serotherapy (eg, ATG). Exclusion criteria included an uncontrolled active infection, abnormal liver function test results (direct bilirubin ≥2 mg/dL, ALT or AST ≥5 times the upper limit of normal), hemodialysis within 7 days prior to enrollment, ventilator support or oxygen supplementation exceeding 40% FiO2 within 14 days prior to enrollment, or the presence of acute GVHD. Patients with a history of allergic reaction to AAT were also excluded. Patients who were low risk at both screening time points were not followed for their outcomes.
Control cohort
The MAGIC database and biorepository contains detailed longitudinal biomarker and clinical data prospectively obtained from patients at centers in North America, Europe, and Asia according to an institutional review board approved protocol at each MAGIC participating center. We identified 441 patients who received an allogeneic HCT at 13 MAGIC centers between 2015 and 2019 and had a high-risk MAP on day 7 post-HCT. We identified an additional 105 patients who had a high-risk MAP on day 14 post-HCT for a total of 546 potential controls. We then created a control cohort of 90 high-risk patients (3:1 ratio of controls to cases) such that (1) control patients met study exclusion criteria (ie, no uncontrolled infections or liver, respiratory, or renal failure) and (2) proportionally matched the case population for age, donor type, HLA match, and GVHD prophylaxis. The cohort was constructed in reverse consecutive order in order to reflect as accurately as possible near-contemporaneous supportive care practices. The control cohort included 30 subjects from MAGIC centers that participated in the clinical trial and 60 subjects from nonparticipating centers (supplemental Table 1). All 30 control subjects from participating centers were transplanted prior to the start of the clinical trial. There were no significant differences in the incidence of SR GVHD (13% vs 15%, P = .99) in control subjects from participating centers compared with nonparticipating centers.
Study design
Screening serum samples were obtained on day 7 (±1 day) and shipped overnight to the central laboratory at Mount Sinai. ST2 and REG3α were analyzed by enzyme-linked immunosorbent assay, as previously described.4,10 The concentrations of ST2 are reported as picograms per milliliter and of REG3α as nanograms per milliliter. The MAP was then calculated as a single value between 0.001 and 0.999 according to the following formula: log[−log(1 − MAP)] = −11.263 + 1.844(log10ST2) + 0.577(log10REG3α).10 All samples were assayed in triplicate and simultaneously with 3 quality control (QC) human serum samples with 3 different known concentrations of ST2 and REG3α that produce MAPs that fall within the range of each published Ann Arbor score.10 Coefficients of variation values must fall within 15% for each sample tested, and results of at least 2 of the 3 QC samples performed for each screening assay must fall within established values for both ST2 and REG3α biomarkers for the results to be recorded; if not, the assays are repeated until at least 2 of the 3 QC samples fall within acceptable parameters. Levey Jennings plots are then created and QC data recorded for every enzyme-linked immunosorbent assay result and calculated MAP. The validated MAP threshold of >0.16 was used to determine risk category, and results were communicated to centers the same day samples were received. Patients who were identified as high risk on day 7 were eligible to enroll provided no exclusion criteria developed while awaiting screening results. Patients who were low risk on day 7 and who met all other eligibility criteria were re-screened on day 14 (±1 day). Patients who were identified as high risk on day 14 were also eligible to enroll on the clinical trial provided no exclusion criteria developed while awaiting screening results. Patients who were identified as low risk on both day 7 and day 14 were ineligible to enroll.
Treatment
Treatment with AAT (Glassia) was initiated within 2 days of confirmation of the high-risk MAP score. AAT was administered intravenously at a loading dose of 90 mg/kg (study day 0), followed by twice weekly doses of 45 mg/kg for 15 more doses (totaling 16 doses over 8 weeks). We reasoned that preemptive treatment of asymptomatic patients would be possible with a lower dose of AAT than that used to treat SR GVHD. We therefore selected a dose that was 75% of the dose used to treat SR GVHD.18 The 8-week treatment duration was selected to provide protection during the period when GVHD most frequently develops.19 Patients continued therapy until any of the following events occurred: completion of all planned doses, development of SR GVHD, change in clinical course rendering further treatment unacceptable, or voluntary withdrawal from treatment. Delayed treatments were allowed to be rescheduled but all infusions needed to be completed within 10 weeks from the first dose. The initial treatment of GVHD was 2 mg/kg prednisone. Additional systemic immunosuppression was initiated at the discretion of the treating physician.
Correlative studies
Serum specimens were collected weekly through study day 28 and at study day 56. Additional serum specimens were collected at GVHD onset and weekly thereafter for 4 weeks.
GVHD data collection
Acute GVHD was staged according to published MAGIC guidelines.20 GVHD staging and medications were recorded weekly through day 100 post-HCT and for 4 weeks after the start of treatment of GVHD, whichever came later. The data collection schedule was the same for both clinical trial subjects and control subjects, and end points were assessed identically for both cohorts. Staff at each site were required to demonstrate proficiency in the application of MAGIC GVHD staging guidelines before entering data, all data were centrally reviewed, and all inconsistencies resolved within 6 months of data entry.
Statistical methods
The primary end point was the incidence of SR GVHD within 100 days after HCT. SR GVHD was defined as the development of GVHD that either (1) did not respond within 28 days of systemic steroid treatment of acute GVHD or (2) required initiation of additional systemic immunosuppression within 28 days of systemic steroid treatment.4,21,22 A complete response was defined as complete resolution of GVHD symptoms in all 3 target organs. Partial response was defined as an improvement in stage in all organs with GVHD involvement without complete resolution of symptoms. Relapse and death were considered competing risks for GVHD, and relapse was considered a competing risk for NRM.
Secondary end points included grade II-IV, grade III-IV, and stage 3 or 4 lower GI GVHD, NRM, relapse, and overall survival (OS). Relapse and death were considered competing risks for GVHD, and relapse was considered a competing risk for NRM. All cumulative incidences were compared by Gray’s tests.23 OS was calculated by the Kaplan-Meier method and compared by log-rank test. Incidences were compared by Fisher’s exact test. MAPs were compared by the Mann-Whitney U test.
Safety end points included tabulation of adverse events and serious infections. Adverse events were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Serious infections were defined as grade 3 systemic infections using Blood and Marrow Transplant Clinical Trial Network criteria.24
For this pilot study, we needed a sample size sufficient to determine if use of the MAP as an inclusion criteria for a multicenter preemptive treatment trial was feasible and to provide guidance as to whether a larger study testing AAT as preemptive treatment should be performed. Pilot studies are not intended to provide conclusive evidence, and type 1 errors up to 0.25 are acceptable.25 Based on these criteria,we calculated that a 30-patient sample size provided 85% power to detect a 13% improvement in the historical incidence of SR GVHD from 28% to 15% using a 1-sided exact test with a type 1 error of 0.23.
Subject screening and enrollment
A consort diagram depicting the screening process is shown in Figure 1. At day 7, 190 patients were screened, and 20 were high risk by MAP. One patient with a high MAP on day 7 developed respiratory failure the next day and was excluded. Of the 170 patients who were low risk at day 7 and monitored for another week, 7 were excluded because they developed GVHD (n = 3) or respiratory failure (n = 3) or died (n = 1). Of the remaining 163 patients who were rescreened on day 14, 13 additional patients were high risk by MAP. Two of these patients were excluded because they developed either GVHD (n = 1) or respiratory failure (n = 1) by the time MAP results were reported the following day. Altogether, 33 out of 190 patients (17%) had high MAPs early post-HCT, and 30 met all eligibility criteria and enrolled in the clinical trial.
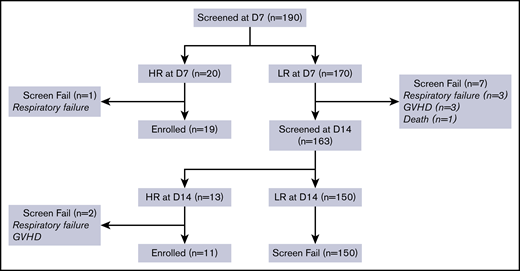
Screening and enrollment of trial patients. D, day; HR, high risk; LR, low risk.
Screening and enrollment of trial patients. D, day; HR, high risk; LR, low risk.
Results
The characteristics of the subjects and controls are shown in Table 1. The control cohort was well matched to the study subjects with the exception that fewer controls received tacrolimus and sirolimus as GVHD prophylaxis compared with study subjects.
Safety and feasibility
Significant (grade ≥3) treatment-emergent adverse events are shown in supplemental Table 2. The most common adverse events observed during AAT therapy were pulmonary complications, including acute respiratory distress syndrome (n = 2), idiopathic pneumonia syndrome (n = 2), diffuse alveolar hemorrhage (DAH) (n = 1), and pulmonary edema (n = 1). AAT therapy was considered related to DAH in 1 subject who experienced the complication twice within hours of consecutive infusions. However, the incidence of respiratory failure was similar in study subjects and controls (13% vs 19%, P = .59).
Four subjects (13%) experienced reactions known to occur with AAT infusions, including grade 1 pruritus, grade 2 supraventricular tachycardia, grade 3 urticaria, and grade 3 facial edema. One subject discontinued AAT infusions due to recurrent episodes of urticaria and facial edema despite premedication with diphenhydramine and acetaminophen; the other 3 subjects experienced no additional reactions with subsequent infusions.
Therapy with AAT consisted of twice-weekly infusions for 8 weeks with planned early cessation in patients requiring additional treatment of SR GVHD. Treatment was begun at a median of 1 day (range, 0-3 days) from confirmation of the high-risk MAP score. Twenty-four patients (80%) received all protocol defined doses of AAT. Six subjects discontinued therapy early due to infusion reaction (n = 1), adverse events (acute respiratory distress syndrome, n = 2; DAH, n = 1), and physician discretion (n = 2).
Clinical outcomes
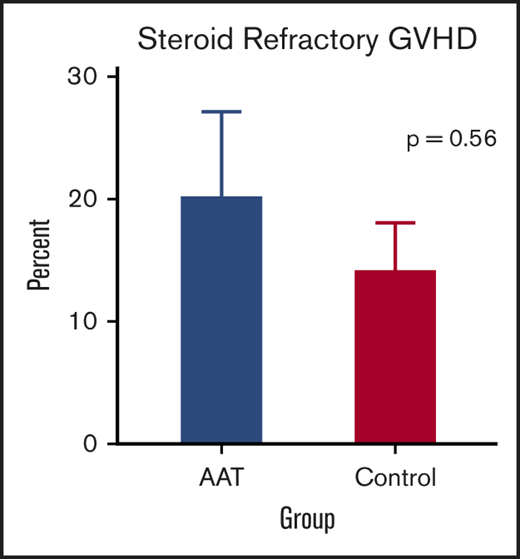
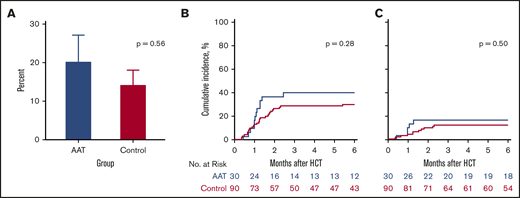
The primary end point for the trial was the development of SR GVHD by day 100 post-HCT. When we compared study subjects to controls for GVHD outcomes, patients treated with AAT were as likely to develop SR GVHD by day 100 as control patients (20% vs 14%, P = .56) (Figure 2A). A more broad definition of SR GVHD includes patients whose GVHD worsened within 3 days or failed to respond within 7 days of treatment, regardless of response at day 28. We identified more SR GVHD using the more broad definition, but the differences between AAT cases and controls was not statistically significant (27% vs 29%, P = .99). Furthermore, treatment with AAT did not reduce the 6-month cumulative incidence of significant GVHD (grade II-IV) requiring systemic treatment (40% vs 30%, P = .28) (Figure 2B) or severe (grade III-IV) GVHD (17% vs 12%, P = .50) (Figure 2C). Treatment of patients with a high-risk MAP using AAT also did not reduce the proportion of patients who developed severe stage 3 or 4 lower GI GVHD by day 100 (13% vs 9%, P = .49) (supplemental Figure 1).
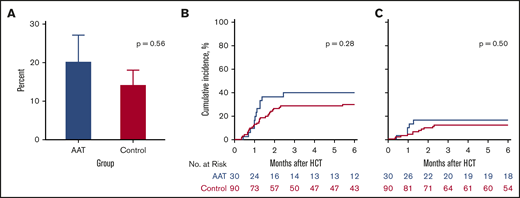
GVHD-related outcomes. (A) Percentage of SR GVHD in AAT-treated patients (blue; 20%) and controls (magenta; 14%) by day 100. (B) Six-month cumulative incidence of grade II to IV GVHD treated with systemic steroids in AAT-treated patients (40%) and controls (30%). (C) Six-month cumulative incidence of grade III or IV GVHD in AAT-treated patients (17%) and controls (12%).
GVHD-related outcomes. (A) Percentage of SR GVHD in AAT-treated patients (blue; 20%) and controls (magenta; 14%) by day 100. (B) Six-month cumulative incidence of grade II to IV GVHD treated with systemic steroids in AAT-treated patients (40%) and controls (30%). (C) Six-month cumulative incidence of grade III or IV GVHD in AAT-treated patients (17%) and controls (12%).
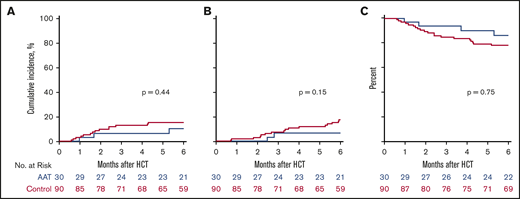
Treatment with AAT also did not significantly decrease the 6-month cumulative incidence of NRM in study subjects compared with controls (10% vs 16%, P = .44) (Figure 3A). All causes of NRM are summarized in supplemental Table 3. Treatment with AAT did not significantly affect the 6-month cumulative incidence of relapse compared with controls (7% vs 18%, P = .15) (Figure 3B) and, as a result, 6-month OS was similar between the 2 groups (86% vs 78%, P = .75) (Figure 3C).
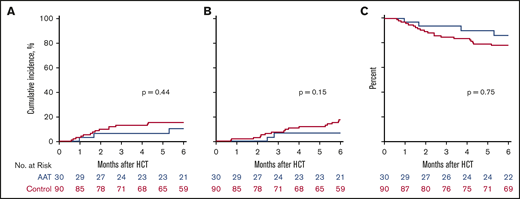
NRM, relapse, and OS. (A) Six-month cumulative incidence of NRM in AAT-treated patients (blue; 10%) and controls (red; 16%). (B) Six-month cumulative incidence of relapse in AAT-treated patients (7%) and controls (18%). (C) Six-month OS in AAT-treated patients (86%) and controls (78%).
NRM, relapse, and OS. (A) Six-month cumulative incidence of NRM in AAT-treated patients (blue; 10%) and controls (red; 16%). (B) Six-month cumulative incidence of relapse in AAT-treated patients (7%) and controls (18%). (C) Six-month OS in AAT-treated patients (86%) and controls (78%).
Correlative studies
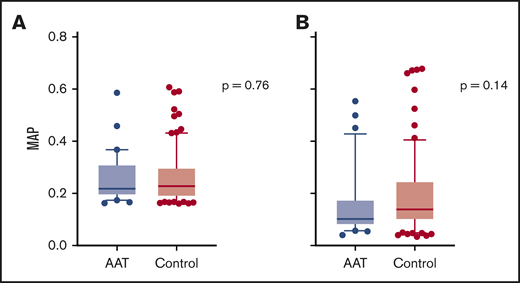
We have shown that the MAP can be used as a monitoring biomarker for the treatment of GVHD.26 At baseline, there was no difference in MAPs between study subjects and controls (median 0.22 vs 0.23, P = .76) (Figure 4A). The MAP declined in both study subjects and controls by a similar amount, and there was no significant difference in the MAPs at day 28 between the 2 groups (median 0.10 vs 0.14, P = .14) (Figure 4B). Thus, AAT treatment of high-risk patients did not alter the subsequent development of GVHD by either clinical or laboratory parameters.
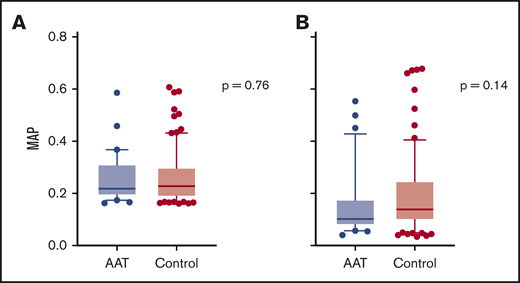
MAP at study entry and day 28 post-HCT. (A) Median MAP at study entry (day 7 or 14 post-HCT) in AAT-treated patients (0.22, n = 30) and controls (0.23, n = 90). (B) Median MAP at day 28 post-HCT in AAT-treated patients (0.10, n = 30) and controls (0.14, n = 82). Box and whisker plots show median MAP with whiskers extending from the 10th to the 90th percentile. Values outside this range are drawn as individual points.
MAP at study entry and day 28 post-HCT. (A) Median MAP at study entry (day 7 or 14 post-HCT) in AAT-treated patients (0.22, n = 30) and controls (0.23, n = 90). (B) Median MAP at day 28 post-HCT in AAT-treated patients (0.10, n = 30) and controls (0.14, n = 82). Box and whisker plots show median MAP with whiskers extending from the 10th to the 90th percentile. Values outside this range are drawn as individual points.
Discussion
Given the poor prognosis of SR GVHD, preemptive treatment prior to the onset of symptoms of patients identified as high risk for this complication is an attractive strategy to improve long-term outcomes. In this multicenter proof-of-concept study, we used the MAP based on serum biomarkers to identify high-risk patients on day 7 or 14 post-HCT and initiate preemptive AAT treatment within 2 days of biomarker confirmation. A strength of this study was its use of a near-contemporaneous, matched control population that provided a more accurate estimate of outcomes of standard treatment than historical controls who received transplants up to 10 years ago. Controls were obtained from the MAGIC database, which contained a large cohort of patients whose natural history was prospectively documented and who had provided serum samples that were analyzed for matching purposes. We applied the study eligibility criteria to controls, thereby reducing the potential to bias comparisons in favor of the study population. Both the study and control populations were treated at centers that had harmonized their practices to score consistently complicated end points such as GVHD staging, further enhancing the reliability of the comparisons. The incidence of SR GVHD and 6-month NRM in the control population (14% and 16%, respectively) was approximately half that of the historical rate and likely reflects the use of controls who had to meet study inclusion and exclusion criteria as well as the recent trend to less severe GVHD and NRM as also reported by others.27 One limitation of the matching process was that fewer control subjects received sirolimus as part of their GVHD prophylaxis regimen. However, the incidence of SR GVHD in high-risk patients (cases or controls) was not significantly different between patients whose GVHD prophylaxis regimen included sirolimus or not (21% vs 13%, P = .43). The processes used to create the control population thus were robust and lent confidence to the comparisons.
This study found that AAT therapy was generally safe and well tolerated, with few infusion reactions and toxicities. Serious pulmonary complications were common (20%) but respiratory failure developed in 2 out of 33 patients (6%) before any AAT had been administered in this high-risk population, and patients treated with AAT were not more likely to develop respiratory failure than controls. A biological linkage between GI damage and idiopathic pneumonia syndrome has previously been observed in experimental allogeneic HCT.28,29 Taken together, these observations suggest that pulmonary adverse events were more likely related to complications of allogeneic HCT itself than AAT treatment, and thus, we do not consider these results to alter the safety profile of AAT.
We chose to investigate AAT based on its excellent safety profile and its efficacy as a treatment of SR GVHD. The study found that subjects were as likely to develop SR GVHD as control patients, and no significant differences between groups were observed in any of the secondary end points, including the cumulative incidence of grade III or IV GVHD, stage 3 or 4 lower GI GVHD, NRM, and OS. The MAP on day 28 (after ≥4 infusions of AAT) was the same in study and control patients, supporting the lack of biologic effect of AAT on GI crypt damage. A major limitation of this study is that we used a lower dose of AAT than used in prior studies for SR GVHD. We did not monitor AAT serum levels, and it is therefore possible that higher AAT doses would have been more effective. An ongoing multicenter randomized trial of AAT as primary treatment of GVHD (#NCT04167514) using a higher dose of AAT than that used in this study is currently being studied for newly diagnosed GVHD, and results from that trial will be important to determine the safety and efficacy of AAT in that setting. It is also possible that a different intervention would have produced better results than those observed in this trial. Other possible approaches designed to repair GI crypt damage such as interleukin-22, RIPK1 inhibitors, or R-spondin1 might prove more efficacious as preemptive treatments.30-33
There have been few prior attempts to preempt the development of severe GVHD with an intervention in the early post-HCT period due to the lack of a reliable method to risk stratify patients in real time and the lack of an effective, nontoxic intervention. One multicenter trial used blood urea nitrogen and serum bilirubin levels on day 7 post-HCT to identify high-risk patients and randomized them to receive either preemptive ATG or no treatment. Although treatment with ATG resulted in less severe GVHD, there was no corresponding improvement in NRM.34 The current study is the first to test such a preemptive strategy using the real-time assessment of GVHD biomarkers as eligibility criteria. Although AAT treatment did not improve outcomes, the ability to enrich study populations for patients at high risk for severe GVHD nevertheless represents an important advance. Physicians and patients are more willing to accept the risks of a novel, more intensive treatment when the risk of no intervention is high.35 A risk-based strategy thus provides a personalized approach and avoids the lack of specificity inherent to intensified GVHD prophylaxis for all patients. Furthermore, early identification of high-risk patients provides an opportunity to intervene before irreversible damage has occurred. In the future, the use of biomarker-based assessment of risk may be more practical at the time of GVHD diagnosis, when 40% of patients are high risk,26 rather than in the first 2 weeks after HCT, when only 15% of the patients are high risk and outcomes are better overall. In this context, biomarkers will aid in the choice between less intensive and more intensive treatments.
All requests for data sharing should be sent to the corresponding author (john.levine@mssm.edu).
Acknowledgments
The authors thank the patients, their families, and the research staff for their participation.
This work was supported by the National Institutes of Health, National Cancer Institute (grants P01CA03942 and P30CA196521), the Deutsche Jose Carreras Leukämie Stiftung e.V. (grant DJCLS 01 GvHD/2016), and Kamada. Drug was supplied by Kamada.
Authorship
Contribution: U.Ö., J.L.M.F., and J.E.L. conceived and designed the study; F.A., J.B., K.B.-D., U.B., H.K.C., Z.D., A.M.E., I.G., S.G., E.O.H., W.J.H., E.H., S.K., C.L.K., P.M., R.N., R.R., W.R., K.S.S., R.Y., and J.E.L. collected and reviewed the clinical data; S.C.G., K.B.-D., G.M., and S.M.K. performed the laboratory analyses; S.C.G., U.Ö., and J.-Y.L. performed the statistical analyses; S.C.G., Z.D., Y.-B.C., J.L.M.F., and J.E.L. wrote the report; and all authors critically reviewed and edited the final report.
Conflict-of-interest disclosure: U.Ö., J.L.M.F., and J.E.L. are co-inventors on a GVHD patent and receive royalties. U.Ö. receives consultancy fees from AffyImmune Therapeutics. J.E.L. receives consultancy fees from Incyte, Mesoblast, OncoImmune, and Talaris and research funding from Biogen, Incyte, and Mesoblast. Z.D. receives consultancy fees from Syndax Pharmaceuticals and research funding from Incyte and Regimmune. Y.B.C. receives consultancy fees from Takeda, Magenta, Daiichi, AbbVie, Equilium, Xenikos, and Celularity. P.M. receives consultancy fees from Sobi and Bellicum. R.N. receives consultancy fees from Viracor. The remaining authors declare no competing financial interests.
Correspondence: John E. Levine, Icahn School of Medicine at Mount Sinai, 1 Gustave Levy Pl, Box 1410, New York, NY 10029; e-mail: john.levine@mssm.edu.