Key Points
Patients with MYD88WT WM treated with zanubrutinib achieved a 50% major response rate (including 27% VGPRs) and 18-month PFS rate of 68%.
The safety profile of zanubrutinib was consistent with previous studies in WM, including a low incidence of cardiovascular toxicity.

Abstract
Patients with Waldenström macroglobulinemia (WM) lacking activating mutations in the MYD88 gene (MYD88WT) have demonstrated relatively poor outcomes to ibrutinib monotherapy, with no major responses reported in a phase 2 pivotal study. Zanubrutinib is a novel, selective Bruton tyrosine kinase (BTK) inhibitor designed to maximize BTK occupancy and minimize off-target activity. The ASPEN study consisted of a randomized comparison of zanubrutinib and ibrutinib efficacy and safety in patients with WM who have the MYD88 mutation, as well as a separate cohort of patients without MYD88 mutation (MYD88WT) or with unknown mutational status who received zanubrutinib. Results from the latter single-arm cohort are reported herein. Efficacy endpoints included overall, major and complete (CR) or very good partial response (VGPR) rates, progression-free survival (PFS), duration of response (DOR), and overall survival (OS). Twenty-eight patients (23 relapsed/refractory; 5 treatment-naïve) were enrolled, including 26 with centrally confirmed MYD88WT disease and 2 with unknown MYD88 mutational status. At a median follow-up of 17.9 months, 7 of 26 MYD88WT patients (27%) had achieved a VGPR and 50% a major response (partial response or better); there were no CRs. At 18 months, the estimated PFS and OS rates were 68% and 88%, respectively, while the median DOR had not been reached. Two patients discontinued zanubrutinib due to adverse events. Treatment-emergent hypertension, atrial fibrillation, and major hemorrhages were reported in 3, 1 and 2 patients (including 1 concurrent with enoxaparin therapy), respectively. Results of this substudy demonstrate that zanubrutinib monotherapy can induce high quality responses in patients with MYD88WT WM. This trial is registered on www.clinicaltrials.gov as NCT #03053440.
Introduction
Waldenström macroglobulinemia (WM) is a B-cell malignancy characterized by bone marrow infiltration with monoclonal, immunoglobulin M (IgM)–secreting, lymphoplasmacytic cells that exhibit constitutive activation of the B-cell receptor signaling complex, of which Bruton tyrosine kinase (BTK) is a critical component.1-3 Activating mutations involving the adaptor protein MYD88, almost all involving the substitution of proline for leucine at amino acid position 265 (MYD88MUT), have been reported in >90% of patients,1 with 30% to 40% reporting an additional frameshift or nonsense mutation in the carboxy-terminal (regulatory) domain of the chemokine receptor, CXCR4 (CXCR4WHIM). The presence of one or both mutations impacts clinical presentation, prognosis and response to targeted therapies, in particular, BTK inhibitors.4 The first-in-class BTK inhibitor, ibrutinib, has demonstrated significant activity in patients with WM. In a phase 2 study of 63 ibrutinib-treated patients with relapsed/refractory (R/R) WM and unselected MYD88 mutational status, the major response rate (MRR; partial response or better) was 78%, including 27% of patients with very good partial responses (VGPRs) after a median treatment duration of 47 months,5 and in a companion study of 30 treatment-naïve (TN) patients (all with MYD88MUT disease) the MRR was 83%, including 20% of patients with VGPRs, after a median treatment duration of 13.4 months.6
In contrast to the favorable outcomes observed in patients with MYD88MUT disease, results for ibrutinib-treated patients with wild-type MYD88 (MYD88WT) tumors, while limited, have been poor. Of 5 patients with MYD88WT disease enrolled to the above-mentioned phase 2 study, none achieved a major response.7 The median progression-free survival (PFS) for this subset of patients was only 21 months, compared with 45 months and not reached for patients with MYD88MUT/CXCR4WHIM and MYD88MUT/CXCR4WT disease, respectively.5 A single patient with MYD88WT enrolled in the iNNOVATE substudy (ibrutinib monotherapy) also did not achieve a major response.8 In a study of a more selective BTK inhibitor, acalabrutinib, 9 (64%) of 14 patients with MYD88WT disease achieved a partial response with no VGPRs or complete responses (CRs).9 Genomic studies in patients with MYD88WT disease revealed important differences from those with MYD88MUT disease, most notably, the presence of somatic mutations that activate NFκB downstream of Myddosome-mediated BTK activation.4,10
The number of studies comparing responses between MYD88WT and MYD88MUT patients to immunochemotherapy combinations are limited. Despite comparable MRRs achieved in both MYD88WT and MYD88MUT patients by immunochemotherapy combinations (with or without BTKi), the median PFS estimated for WM patients with MYD88WT compared less favorably to MYD88MUT with the caveat of small sample sizes.11-13
Zanubrutinib is a novel, potent BTK inhibitor that exhibits sustained BTK inhibition in both peripheral blood mononuclear cells and target tissues at the recommended dose of 160 mg twice daily.14-16 In a phase 1/2 study of patients with various B-cell malignancies, 45% of 73 patients with WM who were treated with zanubrutinib achieved a VGPR or CR and 82% achieved a major response after a median follow-up of 30.3 months.17 Among patients with known MYD88WT disease, 5 (62.5%) of 8 achieved a major response, including one patient who achieved a CR. These preliminary data suggested that zanubrutinib could be a favorable therapeutic option for patients with both MYD88MUT and MYD88WT disease.
The ASPEN trial is an open-label, multicenter, phase 3 study in patients with WM requiring treatment. Cohort 1 is a randomized (1:1) comparison of ibrutinib and zanubrutinib in patients with MYD88L265P mutated tumors, the results for which are reported separately.18 In light of the relatively poor reported outcomes for ibrutinib-treated patients as well as the aforementioned early positive results for zanubrutinib, patients assessed as having MYD88WT disease or those with unknown/inconclusive MYD88 mutational status were assigned to receive zanubrutinib in a separate single-arm substudy (Cohort 2). Efficacy and safety results in this cohort of patients treated with zanubrutinib are reported herein.
Methods
Study design and patients
Eligible patients had confirmed R/R WM after ≥1 prior line of therapy or were treatment-naïve (TN) and deemed unsuitable for standard immunochemotherapy based on the presence of documented comorbidities and/or risk factors (eg, age, cardiac, renal or pulmonary comorbidities, infection or others). Patients were required to meet at least one criterion for treatment per International Workshop on Waldenström macroglobulinemia (IWWM) guidelines,19 have measurable disease (immunoglobulin M >5 g/L), adequate end-organ function and absolute neutrophil and platelet counts of ≥0.75 × 109/L and ≥50 × 109/L, respectively. Patients with prior BTK inhibitor exposure, disease transformation, active central nervous system (CNS) involvement, clinically significant cardiovascular disease, or who required warfarin or another vitamin K antagonist were excluded. All patients received 160 mg zanubrutinib twice daily in 28-day cycles until disease progression or intolerance. Treatment modifications for toxicity are outlined in supplemental Table 1. Treatment interruption for ≤2 consecutive cycles and/or ≤2 dose reductions were permitted for management of grade 3/4, treatment-related toxicities.
The trial was approved by the independent institutional review board or independent ethics committee at each study site and conducted in accordance with applicable regulatory requirements, the principles of the Declaration of Helsinki and Good Clinical Practice guidelines of the International Conference on Harmonization. All patients provided written informed consent. The ASPEN study is registered at www.clinicaltrials.gov as NCT#03053440.
Assessments
Bone marrow aspiration and biopsy were collected at baseline, Week 48, and as clinically indicated thereafter (including for confirmation of CR). Bone marrow aspirates were evaluated at baseline for the presence of MYD88 and CXCR4 mutations in a central laboratory (NeoGenomics, Aliso Viejo, CA).18,20,21 MYD88 mutational status was analyzed without CD19+ cell selection using a proprietary assay that employs locked oligonucleotides to block amplification of MYD88WT DNA during PCR followed by bidirectional Sanger sequencing of the amplicon.22 This approach captures MYD88 L265P mutation with a ∼0.5% limit of detection. Mutations in CXCR4 were detected using PCR, followed by bidirectional Sanger sequencing of the amplicon in non-CD19+ selected bone marrow aspirates. The lower limit of assay sensitivity allows detection of 10% to 15% mutant alleles in a background of wild-type allele, and detects nonsense, frameshift, and other mutations spanning amino acids L301 to S352. Using this assay, mutations from T318 to S341 were identified in this study, which includes almost the full range of CXCR4WHIM mutations previously reported.23,24
Quantitative serum immunoglobulins (IgM, IgG, IgA), M-paraprotein and β2-microglobulin levels were measured at baseline, the beginning of each cycle until Cycle 12, and every 3 cycles thereafter. Either contrast-enhanced computed tomography (CT) or magnetic resonance imaging (MRI) scans were performed at baseline; patients with extramedullary disease (EMD) had follow-up scans every 3 cycles until Cycle 12 and every 6 cycles thereafter until progression. Electrocardiograms were performed on Day 1 of Cycles 1 and 2, every 4 cycles thereafter and at end of treatment.
Outcomes
As the cohort 2 substudy of ASPEN was exploratory in nature, there was no pre-specified primary efficacy endpoint. Efficacy end points of interest included the proportion of patients who achieved a CR or VGPR, MRR (partial response or better), ORR (minor response or better), and time-to-event endpoints (PFS, duration of response [DOR], and overall survival [OS]), with responses as assessed by independent review (PAREXEL Informatics, Waltham, MA) and study site investigators in accordance with consensus criteria defined by the 6th IWWM (IWWM-6)25 (supplemental Table 2) as well as on the basis of reductions in IgM levels alone. Responses were assessed every 28 days and every 84 days after Cycle 12.
Adverse events (AEs), including AEs of interest (AEI) based on the known toxicity profile for the class of BTK inhibitors (eg, atrial fibrillation/flutter, hemorrhage including major hemorrhage, hypertension, infections including opportunistic infections), second primary malignancies, tumor lysis syndrome and peripheral blood cytopenia, were assessed for severity in accordance with the National Cancer Institute Common Toxicity Criteria, Version 4.03. Verbatim AE descriptions were coded to Medical Dictionary for Regulatory Activity (MedDRA) preferred terms. MedDRA event terms that qualified for inclusion in each category of AEI are summarized in supplemental Table 3.
Statistical analysis
The efficacy analysis set includes the 26 patients with centrally confirmed MYD88WT disease. Summaries of response rates and time-to-event endpoints employed descriptive statistics. Two-sided, 95% CIs for each category of response (CR/VGPR, MRR, and ORR) were estimated using the Clopper-Pearson method. Patients with missing response assessments were considered non-responders. PFS, defined as the time from enrollment until progression or death, was estimated using Kaplan-Meier (K-M) methodology with censoring. Two-sided, 95% CIs for median PFS were estimated using the Brookmeyer and Crowley method.26 PFS rates at selected time points were estimated using the K-M method, with corresponding 95% CIs estimated using Greenwood’s Formula.27 DOR, defined as the time from initial qualifying response until progression or death, was summarized using analysis methods similar to those for PFS. Duration of follow-up for PFS and DOR was estimated using the reverse K-M method. OS was measured as the time from initial study treatment until death.
Results
Patient characteristics and disposition
Between January 2017 and July 2018, 229 patients who met consensus criteria for a diagnosis of WM28,29 were enrolled from 58 study sites. Of these, 28 patients were assigned to cohort 2 based on MYD88 mutational status: 26 had documented MYD88WT disease and 2 had unknown MYD88 mutational status due to insufficient bone marrow aspirate for mutation detection. The median age was 72 years; 12 (43%) patients were >75 years old (Table 1). Twenty-three patients had R/R disease and 5 were TN. Most were in the intermediate- (39%) or high-risk (43%) prognostic category by the WM International Prognostic Scoring System (IPSS)30 and 54% were anemic (hemoglobin ≤ 110 g/L) at baseline. The median times from initial diagnosis to initiation of zanubrutinib were 1.5 years (range, 0.1-12.4) for TN patients and 4.0 years (range, 0.5-20.3) for R/R patients. Most (87%) R/R patients received 1-3 prior lines of therapy. Among R/R patients, 95% had at least one prior therapy including anti-CD20 monoclonal antibodies (rituximab or ofatumumab) and/or an alkylating agent (cyclophosphamide, chlorambucil, bendamustine, ifosfamide, lomustine, melphalan, cisplatin); 73.9% had at least one prior therapy including a corticosteroid (dexamethasone, prednisone, prednisolone, hydrocortisone, methylprednisone). Eleven of 23 patients (48%) had a major response to their last therapy including 1 CR and 3 VGPRs, 7 had no response (SD/PD), and for 4 patients, the best response was unknown. Two patients (4%) had a history of atrial fibrillation, and 10 (36%) reported a history of hypertension.
As of 31 August 2019, the median duration of follow-up was 17.9 months, with 17 (61%) patients continuing study treatment at data cutoff. Six patients discontinued zanubrutinib for progressive disease, 2 due to AEs, and 3 due to investigator decision or withdrawal of consent (supplemental Figure 1). There were no reports of disease transformation at the time of this analysis. Median treatment duration was 16.4 months (18.6 months for TN, 16.3 months for R/R patients). Nineteen (68%) patients (3 TN, 16 R/R) had ≥12 months of zanubrutinib exposure. The median relative treatment intensity was 97% (range, 51-100).
Efficacy
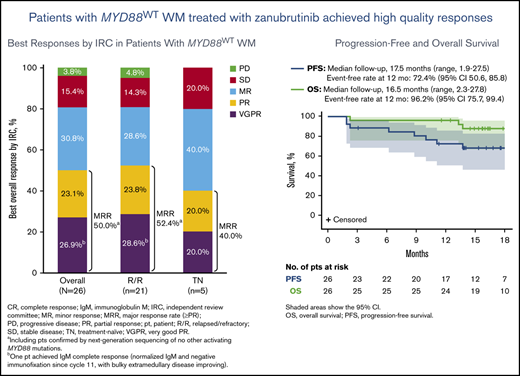
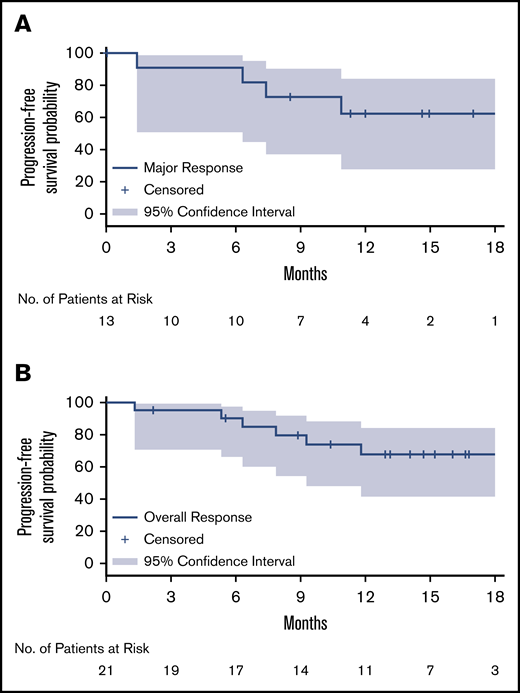
After a median follow-up of 17.9 months, the median PFS and OS for MYD88WT patients treated with zanubrutinib have not been reached. The estimated PFS and OS rates at 18 months were 68% and 88%, respectively (Table 2; Figure 1). Among the 26 patients with confirmed MYD88WT disease, 7 (27%), 13 (50%) and 21 (81%) achieved a VGPR, major response and overall response, respectively, per modified IWWM-6 consensus criteria (supplemental Table 2). No patient achieved a CR (Table 2). Overall response was achieved in 80% and 81% of TN and R/R patients, respectively. The median times to overall response, major response and VGPR were 1.0 month, 2.9 months, and 5.7 months, respectively. The median DOR has not been reached for patients with an overall response, major response or VGPR/CR (Table 2; Figure 2). The concordance rate between IRC- and investigator-assessed best response was 88%. Response rates based on serum IgM reductions alone were similar, with 31%, 54% and 81% of patients having achieved a CR/VGPR, major response and overall response, respectively (supplemental Table 4). The concordance rate for IRC-assessed best response based on IWWM-6 consensus criteria and IgM reductions alone was 92%.
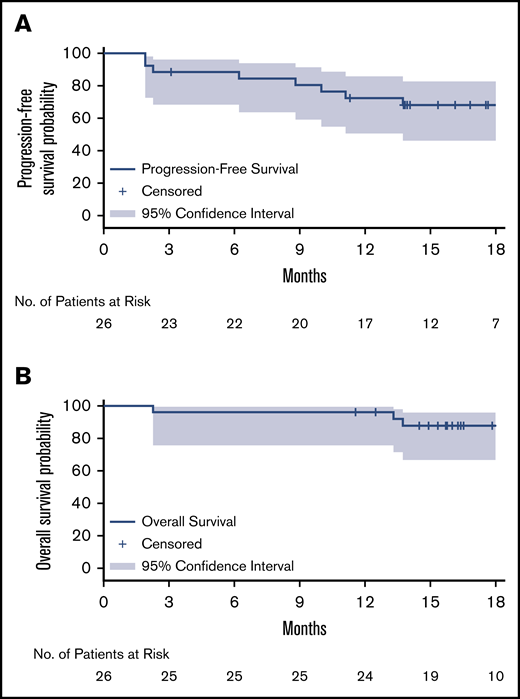
Kaplan-Meier curves for survival probability. PFS (A) and OS (B). Kaplan-Meier distribution for PFS is based on IRC-assessed responses for both relapsed/refractory and treatment-naïve patients.
Kaplan-Meier curves for survival probability. PFS (A) and OS (B). Kaplan-Meier distribution for PFS is based on IRC-assessed responses for both relapsed/refractory and treatment-naïve patients.
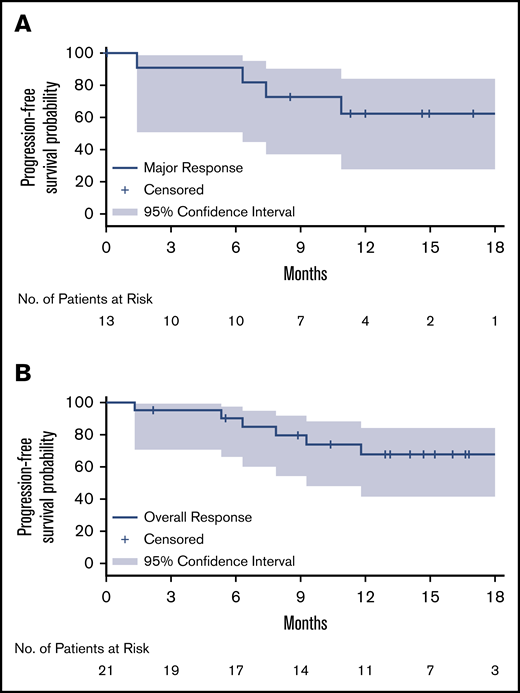
Kaplan-Meier curves for duration of response. Major response (A) and overall response (B). Kaplan-Meier distributions, based on IRC-assessed responses for both relapsed/refractory and treatment-naïve patients.
Kaplan-Meier curves for duration of response. Major response (A) and overall response (B). Kaplan-Meier distributions, based on IRC-assessed responses for both relapsed/refractory and treatment-naïve patients.
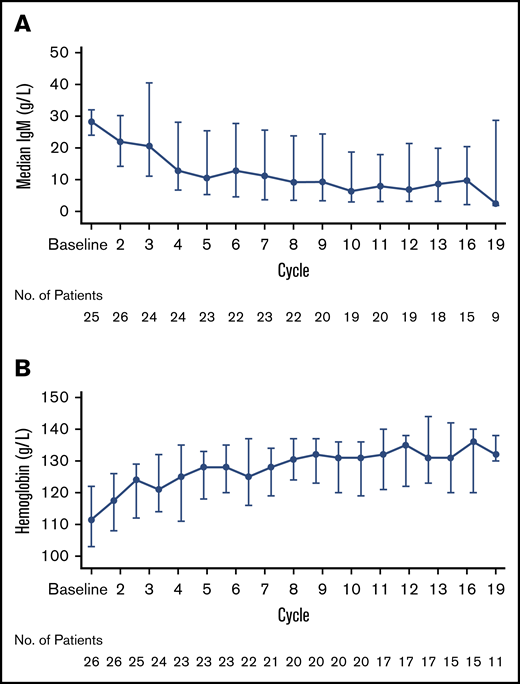
Median maximal reduction in serum IgM levels from baseline was 56% (25th, 75th percentile: 86%, 37%) (Figure 3A) while the median maximal increase in hemoglobin concentration was 19% (25th, 75th percentile: 11%, 24%) (Figure 3B). Seventy-nine percent of patients with CT/MRI evidence of lymphadenopathy and/or splenomegaly at baseline (n = 19) exhibited at least partial reductions in EMD burden.
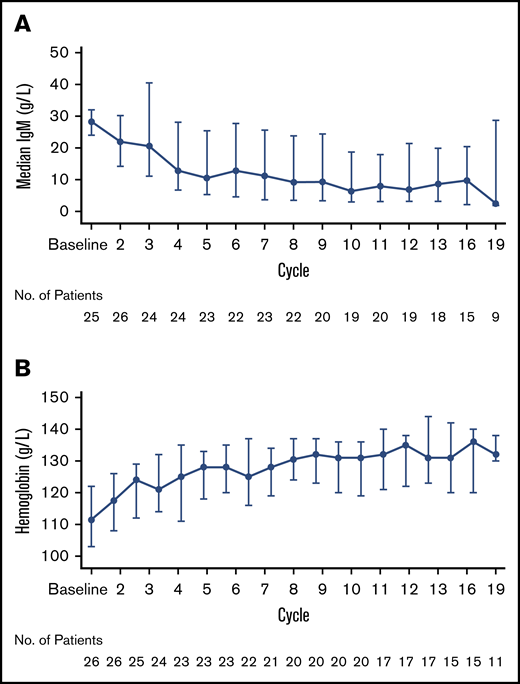
Changes in blood levels. Serum IgM levels (A) and hemoglobin concentrations (B) over time.
Changes in blood levels. Serum IgM levels (A) and hemoglobin concentrations (B) over time.
Safety
Overall, 24 of 28 (85.7%) patients had experienced at least one treatment-emergent AE. AEs reported in ≥20% of patients were diarrhea, upper respiratory tract infection, contusion, pyrexia, and anemia (Table 3). Grade 3 and serious AEs were reported in 18 (64%) and 11 (39%) patients, respectively. Grade 3 AEs reported in at least 5% of patients were neutropenia, anemia, hypertension (11% each), thrombocytopenia, diarrhea, respiratory tract infection, pneumonia and hyponatremia (7% each). The only serious AEs reported in more than 1 patient was pneumonia (in 3 patients), cellulitis and respiratory tract infection (each in 2 patients). At least 1 AEI was reported in 23 (82%) patients. Infections were the most common category of AEIs, with at least 1 infection reported in 75% of patients (Table 3). These were primarily mucosal infections involving the respiratory and urinary tracts, and in 3 of 5 and 4 of 4 patients respectively, were grade 1 or 2. Eight (29%) patients experienced at least 1 Grade ≥3 infection, with pneumonia (n = 2) and respiratory tract infection (n = 2) being most common. No patient reported opportunistic infection. Eleven (39%) patients experienced at least 1 occurrence of hemorrhage; 9 patients with maximum severity of grade 1 (n = 7) or grade 2 (n = 2). Grade 1 contusion (n = 6) and grade 1 hemoptysis (n = 2) were the only bleeding events reported in more than 1 patient. Major hemorrhages (grade ≥3, or central nervous system hemorrhage of any grade) were reported in 2 (7.1%) patients, 1 with periorbital (grade 3) and subdural hematoma/hemorrhage (grade 4) resulting from a fall and in a second patient with bleeding originating from a gastric ulcer (grade 3) in the setting of concurrent enoxaparin therapy. Atrial fibrillation was reported in 1 patient, an 83-year-old female who presented with a grade 1 event coincident with an infection and that resolved within 1 day without intervention or study treatment modification. This patient had no prior history of atrial fibrillation or flutter nor other risk factors apart from her advanced age. Grade 3 hypertension was reported in 3 (11%) patients, 2 of whom had a history of hypertension. Four patients developed second primary malignancies while on study, all of which were skin cancers (3 with basal cell carcinomas and 1 with penile intraepithelial carcinoma).
One patient, an 87-year-old female with a history of hypertension, cryoglobulinemia and vasculitis, developed heart failure 31 days after the last dose of zanubrutinib and died approximately 1 month later from complications thereof. Her death was assessed as unrelated to zanubrutinib. Two additional patients died during the study, one from complications of PD and the other while asleep from unknown cause(s) 11 months after the last dose. Two patients discontinued study treatment of AEs (1 for the aforementioned subdural hematoma/hemorrhage and 1 for grade 3 diarrhea), both assessed as treatment-related. Two patients required dose reductions, 1 for management of grade 1 diarrhea and, in a second patient, coincident with the development of grade 3 pneumonitis on study day 105 and a second dose reduction for grade 2 pneumonia on study day 316.
Discussion
Approximately 3% to 7% of patients with WM harbor tumors that lack an activating mutation in MYD88.4 These patients exhibit a natural history, genomic profiles and response to treatment that vary from those with MYD88MUT disease, leading some to suggest that they represent a unique clinicopathologic entity.4,10,31,32 Retrospective studies indicate that MYD88WT patients exhibit a greater tendency to undergo transformation to aggressive lymphoma; indeed, many exhibit somatic mutations that overlap with those found in diffuse large B-cell lymphoma.4,10,31,32 In at least one relatively large series, MYD88WT patients exhibited shorter overall survival in comparison with patients with MYD88MUT disease, in part due to their greater propensity for disease transformation.32 Almost all patients with MYD88WT WM are also CXCR4WT although in one series, 4 (8.7%) of 46 patients with MYD88WT WM had frameshift mutations in CXCR4.32 In light of their relatively poor outcomes following treatment with single-agent ibrutinib, patients with MYD88WT WM represent an unmet medical need in the era of targeted therapies.
Our study provides clear evidence that single-agent zanubrutinib is effective in patients with MYD88WT WM. Consistent with preliminary results from phase 1/2 studies,17 zanubrutinib induced VGPRs or better in 27% of MYD88WT patients in this substudy (31% based on IgM response alone). This rate is comparable to that reported for R/R and TN patients with MYD88MUT disease treated with zanubrutinib (29% and 26%, respectively).18 Furthermore, the rate of VGPRs reported herein was identical to that reported for the combination of ibrutinib and rituximab in MYD88WT patients (27%) after a median follow-up of 27 months.11 In the current study, 50% (95% CI 30-70) of patients achieved a major response and 81% (95% CI 61-93) achieved an overall response. Taken together, these results suggest that patients with MYD88WT WM treated with zanubrutinib alone could achieve major or overall response rates that are at least similar to those reported for the combination of ibrutinib and rituximab (63% and 81%, respectively in the INNOVATE trial).22 Longer durations of follow-up are required in order to better define the duration of response among WM MYD88WT patients in this substudy. With chemoimmunotherapy, WM patients who achieved VGPR had PFS outcomes indistinguishable from those with a CR.33 If the same holds true for BTKi, the 27% VGPR rate in MYD88WT patients treated with zanubrutinib alone may translate to more favorable PFS compared with other BTKi monotherapies for which VGPR has not been demonstrated.
The molecular basis for the disparity in outcomes between ibrutinib and zanubrutinib in this WM patient subset is unclear since both compounds inhibit BTK via the same mechanism of action, although 100% BTK occupancy sustained over 24 hours with zanubrutinib was demonstrated in paired lymph node biopsies.15 In MYD88MUT WM, autonomous signaling through the Toll/Interleukin-1 Receptor (TIR)/MYD88 complex (or Myddosome) and resulting activation of BTK and NFκB, has a clear role in propagation of the malignant clone.34,35 The importance of Myddosome signaling in MYD88WT patients is less clear cut. Activation of BTK via signaling through the B-cell receptor or possibly other signaling axes might contribute clinically relevant pro-survival signals in MYD88WT patients.3 While published data for patients with MYD88WT WM treated with single-agent ibrutinib are limited,5 it is noteworthy that in a study of acalabrutinib (a more selective BTK inhibitor than ibrutinib), comparable ORRs and MRRs to those reported herein were observed in 14 MYD88WT patients (79% and 64%, respectively, with 95% CI 49–95 and not reported, respectively) although no patient achieved a VGPR.9 To date, the ASPEN study evaluated the largest cohort of WM patients with MYD88WT disease confirmed by central testing and demonstrated VGPR by zanubrutinib monotherapy.
Zanubrutinib was generally well-tolerated in this cohort. The safety profile was consistent with that reported among patients with MYD88MUT disease.18 Infections were the most common category of AEs and were both qualitatively and quantitatively consistent with prior zanubrutinib experience as well as the natural history of WM. The incidence of cardiovascular complications (in particular, atrial fibrillation and hypertension) was comparably low. External precipitating factors contributed to occurrence of the 2 major hemorrhages (ie, head trauma and gastric ulceration/enoxaparin exposure). Peripheral blood cytopenias, while relatively common, were never serious nor did they lead to modification of the study regimen. Quantitatively, other safety metrics such as AEs leading to treatment discontinuation, dose reduction or death were similar to prior experience with zanubrutinib in patients with WM.18
Our study has several limitations, most notably the small number of patients treated and lack of a control arm, although the feasibility of conducting a controlled study given the rarity of MYD88WT WM is questionable. Additionally, the lack of B-cell enrichment in baseline bone marrow aspirates may have precluded the identification of other MYD88 mutations (including non-activating mutations) with low allelic frequency. To our knowledge, the MYD88 mutation detection assay employed in this study with a lower limit of detection of 0.2% to 0.5% had sufficient sensitivity for detection of the most common MYD88 activating mutations.
In summary, this substudy demonstrated that zanubrutinib was, in general, a tolerable and effective treatment option for patients with MYD88WT WM. Further study is needed to clarify the molecular basis for the disparate clinical outcomes observed between ibrutinib-, acalabrutinib- and zanubrutinib-treated patients with MYD88WT WM. Longer follow-up of this cohort will better define the capability of zanubrutinib for disease control in this difficult to manage patient subset.
Presented in part at the 25th Congress of the European Hematology Association, 11-14 June 2020.
All authors had access to original data for the analyses. For the original data, please contact the senior author at mdimop@med.uoa.gr. Individual participant data will not be shared.
Acknowledgments
The authors thank the patients who participated in the study, their supporters, and the investigators and clinical research staff from the study centers.
This study was supported by research funding from BeiGene Inc., US; and medical writing and editorial assistance were funded by BeiGene and provided by Gordon Bray and Bio Connections, LLC.
Authorship
Contribution: All author investigators (M.D., R.G.S., H.-P.L., M.T., M.V., S.O., S.D., R.G.O., G.C., S.M., J.C., J.J.C., M.M., T.S., M.G.M., M.G.G., D.T., P.L.Z., E.A., S.G., A.O., S.R., J.K., A.T., C.B., V.L., J.T., and C.S.T.) collected data; the sponsor confirmed the accuracy of the data and compiled the data for analysis; and all authors reviewed the manuscript and made the decision to submit it for publication and vouch for the accuracy and completeness of the data and analyses and adherence to the trial protocol.
Conflicts-of-interest disclosure: M.D. has received honoraria from and was a consultant/advisor for Amgen, Janssen, Takeda, Celgene, a Bristol-Myers Squibb Company, and Bristol-Myers Squibb. R.G.S. received honoraria from Amgen, Janssen, and Takeda; was a consultant/advisor for Janssen; received research funding from Gilead and Incyte; holds patents, royalties, or other intellectual property from BIOMED 2 Primers, and received travel and accommodations reimbursement from Janssen and Takeda. H.-P.L. owns stock and has ownership in CSL. M.T. received honoraria from Janssen, Gilead Sciences, Takeda, Bristol-Myers Squibb, Amgen, AbbVie, Roche, MorphoSys, and Incyte and was a consultant or advisor for Takeda, Bristol-Myers Squibb, Incyte, AbbVie, Amgen, Roche, Gilead Sciences, Janssen, Celgene, and MorphoSys. M.V. was a consultant or advisor for Janssen and Roche and received travel and accommodations reimbursement from AbbVie and Janssen. S.O. received honoraria from and was a consultant/advisor for AbbVie, Roche, AstraZeneca, Merck, Gilead, Janssen, and Novartis; received research funding from BeiGene, Roche, AstraZeneca, Janssen, Merck, Amgen, and Epizyme; and received travel accomodations and expenses reimbursement from Roche. S.D. participated in speakers’ bureaus for Amgen; received research funding from Janssen and BeiGene; and received travel and accomodations expenses from Janssen. R.G.O. received honoraria from Janssen and Celgene, a Bristol-Myers Squibb Company; was a consultant/advisor for and received travel and accommodations reimbursement from Janssen. G.C. received research funding from BeiGene, Acerta, and Glycomimetics, and received travel and accommodations reimbursement from Glycomimetics. J.J.C. was a consultant/advisor for BeiGene, Janssen, Kymera, Pharmacyclics and Roche, and received research funding from AbbVie, BeiGene, Janssen, Pharmacyclics, and TG Therapeutics. M.M. was a consultant/advisor for Roche and Janssen. T.S. was a consultant/advisor for AstraZeneca, Kite Pharma, Juno Therapeutics, and BeiGene; participated in speakers’ bureaus for Pharmacyclics, Janssen, AstraZeneca, and Seattle Genetics; and received research funding from Pharmacyclics, Juno Therapeutics, BeiGene, AstraZeneca, TG Therapeutics, and Celgene, a Bristol-Myers Squibb Company. M.G.M. participated in speakers’ bureaus for Celgene, a Bristol-Myers Squibb Company, and received travel and accommodations reimbursement from Janssen, Celgene, a Bristol-Myers Squibb Company, Amgen, and Takeda. M.G.G. was a consultant/advisor for Celgene, a Bristol-Myers Squibb Company, and Janssen; and received travel and accommodations reimbursement from Janssen. D.T. received honoraria and research funding from Amgen, Janssen, Roche, and Takeda; and was a consultant/advisor for Roche, Janssen and Amgen. P.L.Z. received honoraria from and participated in speakers’ bureaus for Celltrion, MSD, Gilead, and Astellas; and was a consultant/advisor for Verastem, Kyowa Kirin, and Gilead. A.O. was a consultant/advisor for Celgene, a Bristol-Myers Squibb Company, and Amgen; and participated in speakers’ bureaus for Celgene, a Bristol-Myers Squibb Company, Amgen, and Janssen. S.R. was a consultant/advisor for Janssen, Kite Pharma, Sunesis, and AstraZeneca; received research funding from Janssen; and participated in speakers’ bureaus for Janssen and Roche. A.T. received honoraria from and was a consultant/advisor for Janssen, AstraZeneca, and AbbVie. C.B. received honoraria from, was a consultant/advisor and participated in speakers’ bureaus for Roche, Janssen, Celltrion, and BeiGene; and received research funding from Roche, Janssen, and BeiGene. V.L. received honoraria from AstraZeneca, Roche, Gilead, Amgen, AbbVie, and Janssen; was a consultant/advisor for AstraZeneca, AbbVie, Roche, and Janssen; participated in speakers’ bureaus for AbbVie and Janssen; and received travel and accommodations reimbursement from AbbVie, Roche, and Janssen. J.T. received research funding from BeiGene, Janssen, Celgene, Pharmacyclics, and Roche. W.Y.C. is employed by BeiGene and holds stock/other ownership with BeiGene and Bristol-Myers Squibb. J.M. and J.S. are employed by and hold stock/other ownership with BeiGene. Z.T. is employed by and holds stock/other ownership with BeiGene. A.C. is employed by, holds stock/other ownership with, and received travel and accommodations reimbursement from BeiGene. J.H. is employed by, has a leadership role with, and holds stock/other ownership with BeiGene. C.S.T. is a consultant/advisor for BeiGene, Janssen, Roche, AbbVie, and LOXO; and has received research funding from Janssen, AbbVie, BeiGene, Pharmacyclics, and TG Therapeutics. The remaining authors declare no competing financial interests.
A complete list of the ASPEN investigators appears in the supplemental appendix.
Correspondence: Meletios Dimopoulos, School of Medicine, National and Kapodistrian University of Athens, Alexandra Hospital, 80 Vasilisis Sophias, 11528 Athens, Greece; e-mail: mdimop@med.uoa.gr.