Key Points
With extended follow-up, ibrutinib demonstrated durable clinical benefit in patients with relapsed/refractory MZL.
Biomarker data correlated with clinical outcomes and suggest that ibrutinib’s efficacy may be related to its disruption of NF-κB signaling.

Abstract
Advanced marginal zone lymphoma (MZL) is an incurable B-cell malignancy dependent on B-cell receptor signaling. The phase 2 PCYC-1121 study demonstrated the safety and efficacy of single-agent ibrutinib 560 mg/d in 63 patients with relapsed/refractory MZL treated with prior rituximab (RTX) or rituximab-based chemoimmunotherapy (RTX-CIT). We report the final analysis of PCYC-1121 with median follow-up of 33.1 months (range: 1.4-44.6). Overall response rate (ORR) was 58%; median duration of response (DOR) was 27.6 months (95% confidence interval [CI]: 12.1 to not estimable [NE]); median progression-free survival (PFS) was 15.7 months (95% CI: 12.2-30.4); and median overall survival (OS) was not reached (95% CI: NE to NE). Patients with prior RTX treatment had better outcomes (ORR: 81%; median DOR: not reached [95% CI: 12.2 to NE]; median PFS: 30.4 months [95% CI: 22.1 to NE]; median OS: not reached [95% CI: 30.3 to NE]) vs those with prior RTX-CIT treatment (ORR: 51%; DOR: 12.4 months [95% CI: 2.8 to NE]; PFS: 13.8 months [95% CI: 8.3-22.5]; OS: not reached [95% CI: NE to NE]). ORRs were 63%, 47%, and 62% for extranodal, nodal, and splenic subtypes, respectively. With up to 45 months of ibrutinib treatment, the safety profile remained consistent with prior reports. The most common grade ≥3 event was anemia (16%). Exploratory biomarker analysis showed NF-κB pathway gene mutations correlated with outcomes. Final analysis of PCYC-1121 demonstrated long-term safety and efficacy of ibrutinib in patients with relapsed/refractory MZL, regardless of prior treatment or MZL subtype. This trial was registered at www.clinicaltrials.gov as #NCT01980628.
Introduction
Marginal zone lymphoma (MZL) is an indolent B-cell malignancy with an estimated incidence of ∼7500 new cases annually in the United States.1 Given the indolent nature of the disease course, patients can live for many years after diagnosis, with median overall survival (OS) durations of >12 years for extranodal MZL and >8 years for both nodal and splenic MZL.1,2 Although patients with localized disease can be treated curatively, advanced MZL is incurable, and treatment approaches generally follow those of the more common follicular lymphoma.3,4 Although the disease course of MZL is similar to that of follicular lymphoma, the differences in biology have treatment implications.5-7 Overall, intermittent treatment, the need for surveillance, and the morbidity of therapy have a psychological, physical, and financial burden.8
First-line treatment options for advanced MZL include anti-CD20 antibody monotherapy, chemotherapy, and combination chemoimmunotherapy.3,4 Most patients will experience relapse after first-line therapy, but until recently the only treatment option for patients with relapsed disease was re-treatment with the same agents.3,4 However, cumulative toxicity, particularly with alkylating agents, can lead to stem cell damage, myelodysplasia, and acute myeloid leukemia with an unfavorable prognosis.9 The estimated incidence of treatment-related myelodysplasia or acute myeloid leukemia ranges from 1% to 6% with 20 years of follow-up after conventional chemotherapy.9
Bruton tyrosine kinase (BTK) is a key component of the B-cell receptor signaling pathway.10,11 Genetic BTK mutations lead to a state of immunodeficiency characterized by a lack of mature B lymphocytes, namely, X-linked agammaglobulinemia (Bruton agammaglobulinemia).12 Therefore, inhibition of the B-cell receptor signaling pathway was anticipated to be therapeutic in B-cell malignancies by impeding migration, proliferation, and survival of tumor cells in B-cell malignancies.10-15 Ibrutinib is a once-daily BTK inhibitor approved in the United States for the treatment of patients with MZL who require systemic therapy and have received at least 1 prior anti-CD20–based therapy.16
PCYC-1121, the phase 2 study leading to the Food and Drug Administration approval of ibrutinib for previously treated MZL, demonstrated the efficacy and safety of single-agent ibrutinib across all subtypes of MZL (ie, extranodal lymphoma of mucosa-associated lymphoid tissue [MALT], nodal, and splenic).17 It was also one of the largest prospective trials conducted in advanced stage MZL. After a median follow-up of 19.4 months in the PCYC-1121 study (primary analysis), the overall response rate (ORR) was 48% by independent review; median duration of response (DOR) was not yet reached, and median progression-free survival (PFS) was 14.2 months. A subgroup analysis demonstrated higher response rates in patients who received only prior rituximab (RTX; 69%) compared with those who had received prior rituximab-based chemoimmunotherapy (RTX-CIT; 46%). Because ibrutinib treatment requires chronic dosing, long-term follow-up is necessary to determine the safety and efficacy of ibrutinib over extended dosing periods.
The final efficacy and safety analysis from PCYC-1121 after a median of 33 months of follow-up is presented here. We analyzed subgroups based on the major MZL subtypes (extranodal, nodal, and splenic) and prior lines of therapy (RTX or RTX-CIT). We also conducted biomarker analyses investigating the correlation of somatic variants with clinical outcomes.
Materials and methods
Study design
PCYC-1121 (#NCT01980628) was a phase 2, multicenter, open-label, nonrandomized trial designed to assess the efficacy and safety of single-agent ibrutinib in patients with relapsed/refractory MZL. Study design details have been previously published.17 In brief, patients aged ≥18 years with histologically confirmed MZL of any subtype who were previously treated with ≥1 anti-CD20–directed treatment and had disease progression or failed to achieve at least a partial response (PR) with their most recent treatment received oral ibrutinib 560 mg once daily until unacceptable toxicity, disease progression, or patient withdrawal. PCYC-1121 was approved by institutional review boards or independent ethics committees and was conducted according to the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines from the International Conference on Harmonization. All patients provided written informed consent.
Safety and response assessments
ORR was evaluated by investigators according to the 2007 International Working Group for Non-Hodgkin Lymphoma criteria.18 Response was assessed via computed tomography or magnetic resonance imaging every 12 weeks from the first dose of ibrutinib. Other key efficacy outcomes were DOR, PFS, and OS. Adverse events (AEs) were reported up to 30 days after last treatment dose and regardless of attribution to study drug. Laboratory abnormalities of clinical significance as determined by the investigator were also reported. Severity of AEs was graded according to Common Terminology Criteria for Adverse Events, version 4.03.
Biomarker analysis
In prespecified exploratory analyses, mutation profiling was performed on all available baseline tumor samples using the cancer-specific ACE Extended Cancer Panel (Personalis, Menlo Park, CA), which includes ∼1400 cancer genes and ∼200 microRNA genes. Associations of somatic variants with known clinical relevance in MZL (including MYD88, A20, CARD11, KMT2D, and NOTCH-2)19-21 to aforementioned efficacy measures of best response, DOR, PFS, OS, and maximum change from baseline in sum of the products of longest diameters (SPD) of tumor lesions were analyzed. Results with correlations of P < .05 are reported; exploratory analyses were not powered for significance.
Statistical analysis
Baseline characteristics and safety were assessed in the intention-to-treat (ITT) population, defined as patients who received at least 1 dose of ibrutinib. Efficacy was assessed in the efficacy population, defined as patients with measurable disease at baseline who received at least 1 dose of ibrutinib. Efficacy outcomes were analyzed descriptively by subgroups based on prior line of therapy (RTX or RTX-CIT) and by MZL subtype (extranodal, nodal, or splenic).
ORR was defined as the proportion of patients with a complete response (CR) or PR and was calculated with the corresponding 2-sided 95% confidence interval (CI). The Kaplan-Meier method was used to estimate time to event end points. For the overall population and subgroup analyses, landmark rates are provided at 33 months and correspond with the median follow-up duration. Change in tumor size was assessed by maximum change in SPDs.
Results
Detailed baseline characteristics of the 63 patients included in the study have been reported previously.17 MZL subtypes were as follows: 32 patients (51%) extranodal, 17 patients (27%) nodal, and 14 patients (22%) splenic. Patients had received a median of 2 prior lines of systemic therapy (range: 1-9). Seventeen patients (27%) had received prior treatment with single-agent RTX only; 40 patients (63%) were treated with prior RTX-CIT, and 6 patients (10%) received other prior therapy (Table 1). At the time of final analysis, the median follow-up duration was 33.1 months (range: 1.4-44.6). Treatment discontinuations during the study were due to progressive disease (PD; n = 23, 37%), AEs (n = 12, 19%), physician decision (8%), withdrawal of consent (6%), and noncompliance (2%). Median duration of treatment was 11.6 months (range: 0.2-44.6). At study closure, 18 patients (29%) were still on therapy, 16 of whom opted to participate in a separate treatment extension study (#NCT03229200). All 63 patients were included in the ITT population, and 60 patients were included in the efficacy population. Three patients had nonmeasurable disease at baseline and were excluded from the efficacy population.
Long-term efficacy
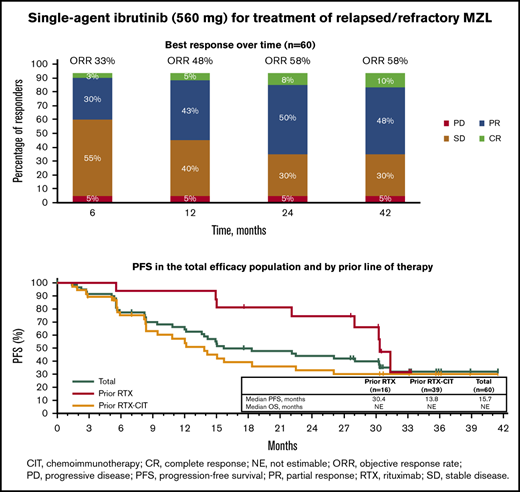
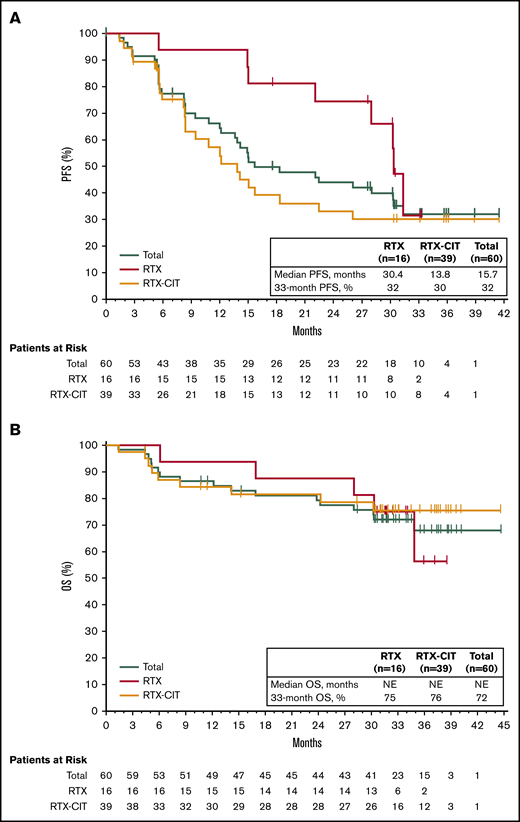
With long-term treatment, the investigator-assessed ORR increased from 53% at the primary analysis to 58% at the final analysis. At the time of the final analysis, 10% of patients (n = 6) had achieved a best response of CR; 48% (n = 29) achieved a PR; 30% (n = 18) had stable disease (SD), and 5% (n = 3) had PD. Four patients who discontinued before the first response assessment were counted as nonresponders (Figure 1A). ORR increased from 48% at 1 year to 58% at 3 years, and CR rates increased from 5% at 1 year to 10% at 3 years (Figure 1B). The first response assessment was at 3 months after treatment initiation, and the median time to initial response was 5.6 months (range: 2.4-22.4). Median DOR from the time of initial response was 27.6 months (95% CI: 12.1 to not estimable [NE]); 48% of patients remained with a response at month 33. Median investigator-assessed PFS was 15.0 months at the primary analysis and 15.7 months (95% CI: 12.2-30.4) in the final analysis, with a PFS rate of 32% at month 33 (Figure 2A). Median OS was not reached, and the OS rate at month 33 was 72% (Figure 2B).
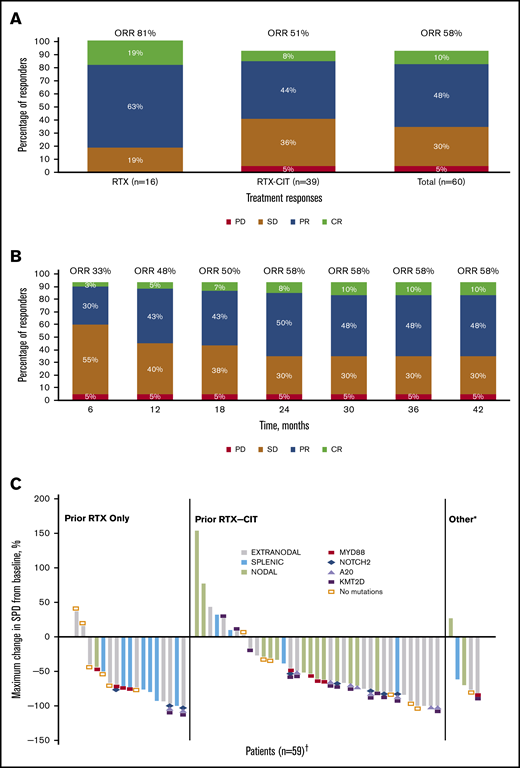
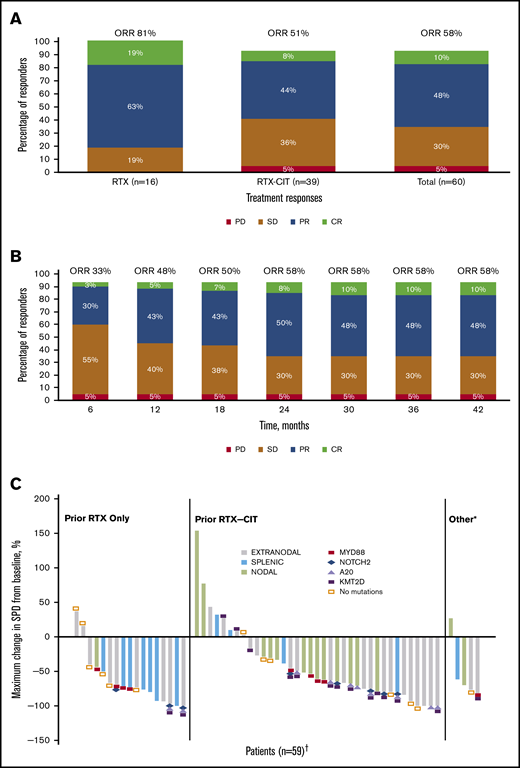
ORR and reduction in lymph node SPD with single-agent ibrutinib treatment. (A) Response rates for total efficacy population and by prior line of therapy. (B) Best response over time in the total efficacy population (n = 60). (C) Change in tumor size by prior line of therapy in the ITT population. *Patients in the “Other” category had prior treatment with both single-agent RTX and chemotherapy or investigational therapies. †Four patients who discontinued before the first response assessment were excluded. PD, progression of disease; SD, stable disease.
ORR and reduction in lymph node SPD with single-agent ibrutinib treatment. (A) Response rates for total efficacy population and by prior line of therapy. (B) Best response over time in the total efficacy population (n = 60). (C) Change in tumor size by prior line of therapy in the ITT population. *Patients in the “Other” category had prior treatment with both single-agent RTX and chemotherapy or investigational therapies. †Four patients who discontinued before the first response assessment were excluded. PD, progression of disease; SD, stable disease.
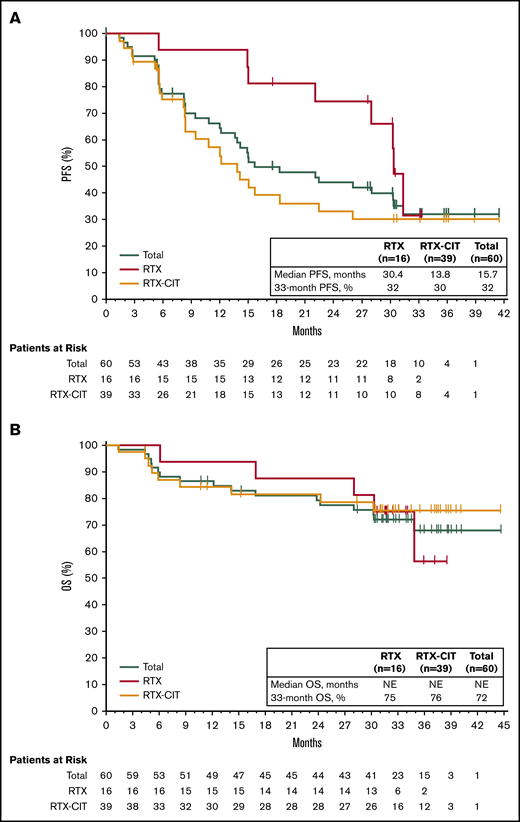
PFS and OS with single-agent ibrutinib treatment. (A) PFS in the total efficacy population and by prior line of therapy. (B) OS in the total efficacy population and by prior line of therapy.
PFS and OS with single-agent ibrutinib treatment. (A) PFS in the total efficacy population and by prior line of therapy. (B) OS in the total efficacy population and by prior line of therapy.
Subgroup efficacy analysis by prior therapy
Of the patients treated with prior single-agent RTX, ORR was 81% with 19% CR (n = 3) and 63% PR (n = 10); 19% (n = 3) had SD, and no patients had PD. Among those treated with prior RTX-CIT, ORR was 51% with 8% CR (n = 3) and 44% PR (n = 17); 36% of patients (n = 14) had SD and 5% (n = 2) had PD (Figure 1A; supplemental Figure 1). A reduction in tumor size was observed in 81% of patients (48/59), including 88% (15/17) treated with prior RTX and 78% (29/37) treated with prior RTX-CIT; 4 patients who discontinued before the first response assessment were excluded (Figure 1C). Patients treated with prior single-agent RTX had a median time to initial response of 2.8 months (range: 2.4-19.4), whereas those treated with prior RTX-CIT had a median time to initial response of 5.6 months (range: 2.7-22.4). Median DOR in patients treated with prior RTX was not reached (95% CI: 12.2 to NE). DOR in those treated with prior RTX-CIT was 12.4 months (95% CI: 2.8 to NE). For patients treated with prior RTX, the DOR rate at 33 months from the time of initial response was NE (95% CI: NE to NE); for those treated with prior RTX-CIT, the DOR rate at 33 months from the time of initial response was 47% (95% CI: 24.4 to 67.3) (Table 2).
Of the patients treated with prior RTX, median PFS was 30.4 months (33-month PFS rate, 32% [95% CI: 6.4-61.5]), whereas median PFS was 13.8 months (33-month PFS rate, 30% [95% CI: 15.8-45.7]) for patients treated with prior RTX-CIT (Figure 2A; Table 2). Median OS for patients treated with prior RTX and RTX-CIT was not reached. OS rates at 33 months were similar regardless of prior line of therapy (prior RTX, 75% [95% CI: 46.3-89.8]; prior RTX-CIT, 76% [95% CI: 58.1-86.5]) (Figure 2B; Table 2).
Subgroup efficacy analysis by MZL subtype
Response rates were substantial across all MZL subtypes. ORRs were 63% (19/30 patients), 47% (8/17), and 62% (8/13) for patients with the extranodal, nodal, and splenic subtypes, respectively (supplemental Figure 1; supplemental Table 1). PFS and OS for the MZL subtypes are shown in supplemental Figures 2 and 3. Median time to initial response ranged from 2.8 months in patients with splenic MZL to 16.6 months in patients with nodal MZL. DOR, PFS, and OS rates at 33 months were generally similar across MZL subtypes (Table 2).
Safety
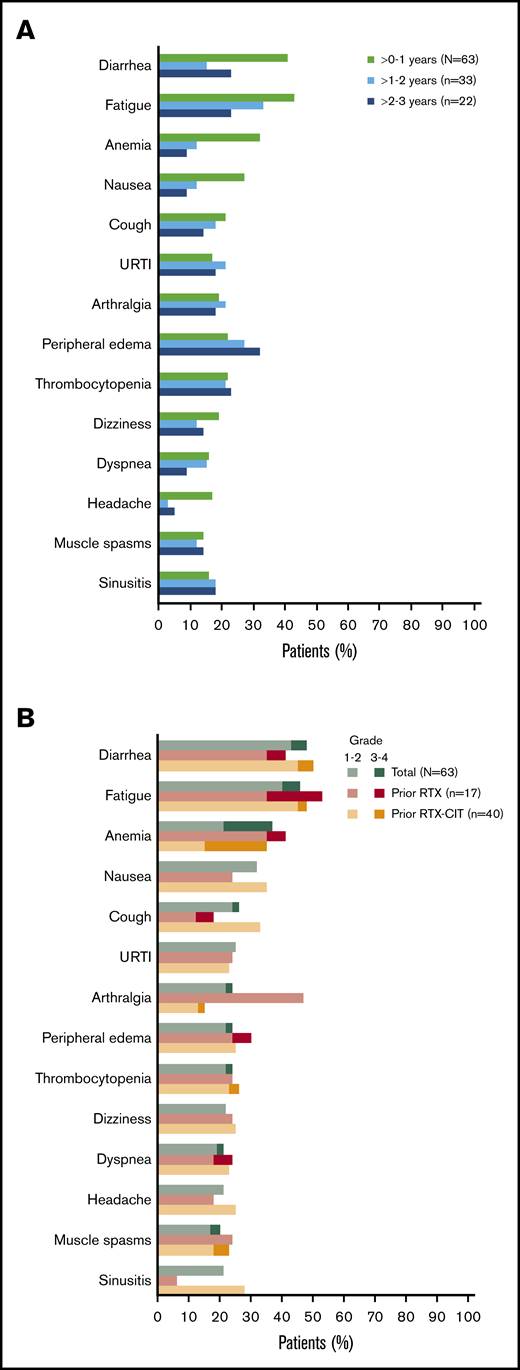
All patients had a treatment-emergent adverse event (TEAE), and 71% (n = 45) had a TEAE of grade 3 or higher. The most common TEAEs of any grade were diarrhea (48%; n = 30), fatigue (46%; n = 29), anemia (37%; n = 23), and nausea (32%; n = 20); the prevalence of the most common TEAEs of any grade generally remained stable or decreased over time (Figure 3A). The most common grade 3 or higher TEAE was anemia (16%; n = 10) (Figure 3B). Overall, the prevalence of grade 3 or higher TEAEs decreased from 59% in years >0 to 1 to 42% in years >1 to 2, and to 36% in years >2 to 3. Grade 3 or higher infections occurred in 22% of patients (n = 14); most of these infections occurred within the first year of treatment (n = 10), with 4 patients experiencing an infection after the first year of treatment. Atrial fibrillation occurred in 8% of patients (n = 5). Of these 5 patients, 3 had atrial fibrillation events within the first year of treatment. All atrial fibrillation events were grade 1/2 in severity, and none required dose modification or discontinuation of ibrutinib. Bleeding events of any grade occurred in 68% of patients (n = 43) and were primarily grade 1/2 (65%; n = 41); major hemorrhage occurred in 3% of patients (n = 2). Serious TEAEs occurred in 46% of patients (n = 29), the most common of which was pneumonia (8%; n = 5). TEAEs (grade 1 to 4) led to discontinuation in 17% of patients (n = 11) and included diarrhea (n = 2); pneumonia (n = 2); and eosinophilic pneumonia, abnormal hepatic function, lymphoma, pneumonitis, pulmonary embolism, rash maculopapular, and rash papular (n = 1 each). Discontinuation occurred primarily within the first year of treatment (10/11 patients). Overall, 5% of patients (n = 3) had TEAEs resulting in death: worsening of disease (n = 1), parainfluenza virus infection leading to multiple organ dysfunction syndrome (n = 1), and cerebral hemorrhage (n = 1). Of these, death due to parainfluenza virus infection was deemed possibly related to ibrutinib. Cerebral hemorrhage occurred in a patient 19 days after ibrutinib discontinuation and was reported as unlikely related to ibrutinib, as the patient received anticoagulation with dalteparin prior to the bleeding event.
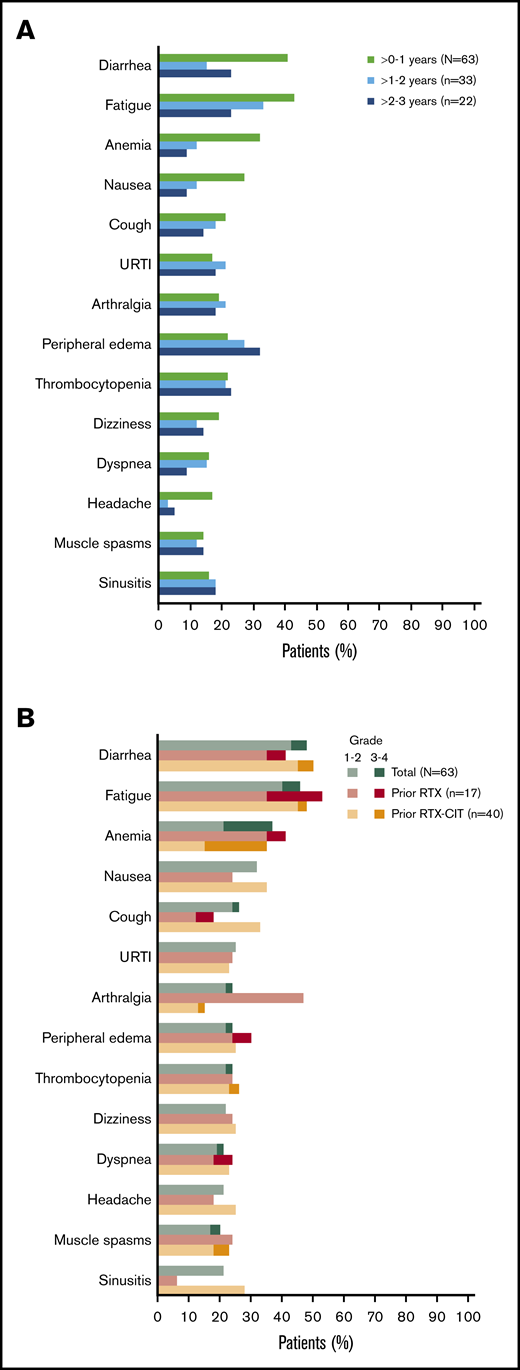
Most common AEs. (A) Prevalence of most common TEAEs* of any grade over time† in the total safety population. (B) Most common TEAEs* in the total safety population and by prior line of therapy. URTI, upper respiratory tract infection. *Occurring in ≥20% of patients overall in the total safety population. †Prevalences of TEAEs beyond 3 years have been excluded because of small sample size (n = 7).
Most common AEs. (A) Prevalence of most common TEAEs* of any grade over time† in the total safety population. (B) Most common TEAEs* in the total safety population and by prior line of therapy. URTI, upper respiratory tract infection. *Occurring in ≥20% of patients overall in the total safety population. †Prevalences of TEAEs beyond 3 years have been excluded because of small sample size (n = 7).
Biomarkers
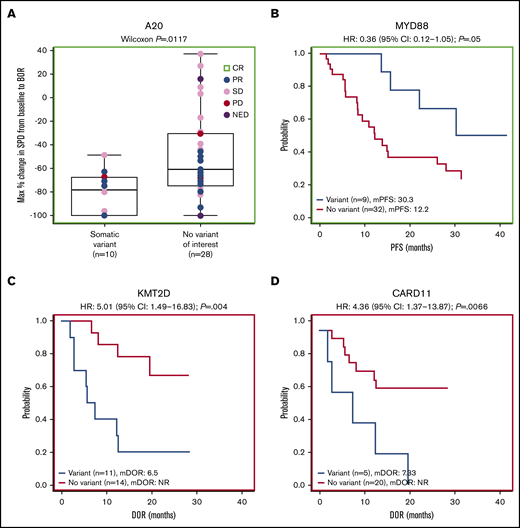
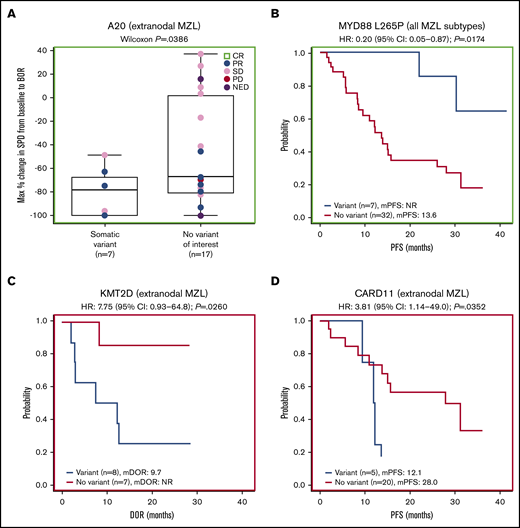
Forty-one patients had baseline targeted DNA-sequence data available for biomarker analyses; in these patients, outcomes data were also available for best response (n = 38), DOR (n = 25), PFS (n = 41), OS (n = 41), and SPD (n = 38). Of the clinically relevant genes tested, mutations of 2 genes correlated with improved efficacy. Among 38 patients with SPD data, those with mutations in A20 (TNFAIP3; n = 10) had significantly greater tumor shrinkage than wild type (P = .0117; Figure 4A). Among 24 patients with the extranodal MZL subtype with SPD data, those with mutations in A20 (n = 7) also had significantly greater tumor shrinkage than wild type (P = .0386; Figure 5A). Among 41 patients with PFS data, those with a mutation in MYD88 (n = 9) had significantly longer median PFS (30.3 months) compared with wild type (12.2 months; hazard ratio [HR]: 0.36) (P = .0500; Figure 4B). PFS was also longer in patients with the MYD88 L265P variant (n = 7) compared with wild type (median PFS not reached vs 13.6 months; HR: 0.20) (P = .0174; Figure 5B).
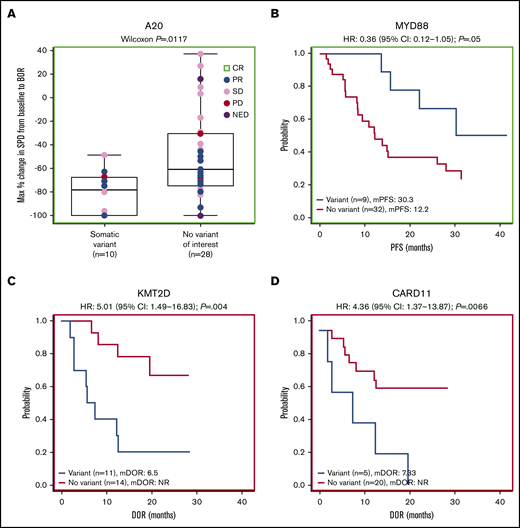
Gene-level mutational analyses correlate to clinical outcomes. Mutations in A20 (A) and MYD88 (B) correlate to favorable prognosis. Mutations in KMT2D (C) and CARD11 (D) correlate to poor prognosis. BOR, best overall response; mDOR, median DOR; mPFS, median PFS; NED, no evidence of disease.
Gene-level mutational analyses correlate to clinical outcomes. Mutations in A20 (A) and MYD88 (B) correlate to favorable prognosis. Mutations in KMT2D (C) and CARD11 (D) correlate to poor prognosis. BOR, best overall response; mDOR, median DOR; mPFS, median PFS; NED, no evidence of disease.
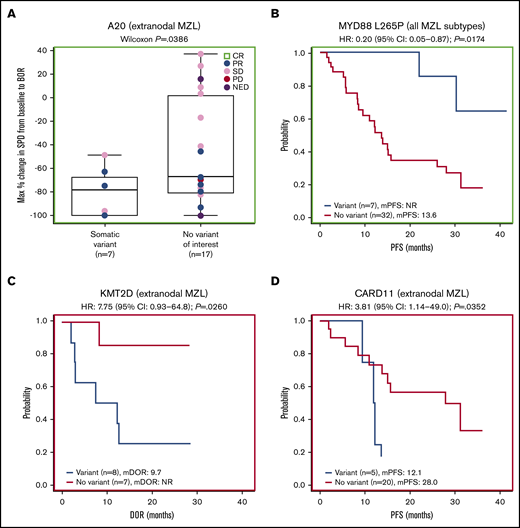
Extranodal subgroup and gene variant analyses. (A) Gene-level analysis for A20 in patients with extranodal MZL. (B) Analysis of the MYD88 L265P variant for all MZL subtypes combined. (C) Gene-level analysis for KMT2D in patients with extranodal MZL. (D) Gene-level analysis for CARD11 in patients with extranodal MZL.
Extranodal subgroup and gene variant analyses. (A) Gene-level analysis for A20 in patients with extranodal MZL. (B) Analysis of the MYD88 L265P variant for all MZL subtypes combined. (C) Gene-level analysis for KMT2D in patients with extranodal MZL. (D) Gene-level analysis for CARD11 in patients with extranodal MZL.
Mutations of 2 genes correlated with poor efficacy. Among 25 patients with available DOR data, those with mutations in KMT2D (n = 11) and CARD11 (n = 5) had significantly worse median DOR (6.5 and 7.3 months, respectively) compared with wild type (not reached for both; HR: 5.01 and 4.36, respectively) (P = .0040, P = .0066; Figure 4C-D). We found significantly worse DOR in patients with extranodal MZL with variants in KMT2D (n = 8 in 15 patients with DOR data), with a median DOR of 9.7 months vs not reached in wild type (HR: 7.75) (P = .0260; Figure 5C), and significantly worse PFS in patients with extranodal MZL with variants in CARD11 (n = 5 in 25 patients with PFS data), with a median PFS of 12.1 months vs 28.0 months in wild type (HR: 3.81) (P = .0352; Figure 5D). In all analyses, NOTCH-2 mutations did not significantly correlate to any of the 5 efficacy measures investigated. Mutations in clinically relevant genes did not correlate with OS.
Discussion
We assessed the long-term efficacy and safety of single-agent ibrutinib in patients with relapsed/refractory MZL. With a median follow-up of 33.1 months in this final analysis of the phase 2 PCYC-1121 study, representing ∼14 months of additional follow-up since the primary analysis, single-agent ibrutinib continues to demonstrate durable clinical benefit in patients with relapsed/refractory MZL. The updated results are consistent across MZL subtypes and clinical characteristics. Response deepened over time: ORR increased from 48% at 1 year to 58% at 3 years, and CR rates increased from 5% at 1 year to 10% at 3 years. After 33 months of follow-up, OS remained not yet reached. Furthermore, responses were durable with a median DOR of 27.6 months, with 48% of patients continuing to respond at 33 months. No new safety signals emerged. Results of the PCYC-1121 study were the basis for the first disease-specific approval in relapsed/refractory MZL. These updated results provide continued evidence of the efficacy and safety of single-agent ibrutinib and support the use of ibrutinib as a chemotherapy-free option for relapsed/refractory MZL.
Immunotherapy (RTX monotherapy) or chemoimmunotherapy with R-CHOP (RTX in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone) or bendamustine plus RTX (BR) are standard first- or second-line treatments for indolent non-Hodgkin lymphomas (NHLs). However, data on the use of these regimens are typically from trials of predominantly follicular lymphoma with small subgroups of MZL patients. Most previous studies published for MZL were conducted in treatment-naive patients who generally have better and more durable responses than patients with relapsed/refractory MZL. Thus, effective treatment for patients with relapsed/refractory MZL has been elusive. Treatment with chemotherapy or chemoimmunotherapy regimens is associated with grade 3/4 myelosuppression, and treatment with multiple lines of therapy is associated with poorer outcomes in subsequent therapy for other B-cell malignancies.22-24 Repetitive treatment with alkylating agents is also associated with myelodysplasia and acute myelogenous leukemia.9 Therefore, chemotherapy-free treatment is an attractive and effective option for patients with relapsed/refractory MZL following immunotherapy.
The results of the subgroup analyses in patients previously treated with either single-agent RTX or RTX-CIT should be viewed alongside other single-agent treatments for indolent NHL, although comparisons are complicated by differences in patient populations across studies. At the time of final analysis in the current study, the ORR for chemotherapy-naive patients treated with prior single-agent RTX was 81% (CR 19%), whereas the ORR for patients with prior RTX-CIT was 51% (CR 8%). These findings are consistent with those of a phase 2 trial conducted in patients with MALT MZL (n = 34), in which single-agent RTX yielded an ORR of 73%, with higher ORR in chemotherapy-naive patients vs those with prior chemotherapy (87% vs 45%).25 ORRs of 85% to 93% have been reported with RTX in combination with chemotherapy in studies that primarily included patients who were treatment- or chemotherapy-naive.26-30 In the relapsed/refractory setting, RTX combined with chemotherapy has demonstrated ORRs of 77% to 93% in patients with MALT MZL, but 29% to 31% of patients developed grade 3/4 cytopenias.31,32 Efficacy has also been observed with phosphatidylinositol-3-kinase δ inhibitors, with ORRs of 47% to 70% in the subsets of patients with relapsed/refractory MZL.33,34 Most recently, a study of lenalidomide combined with RTX vs placebo plus RTX showed ORRs of 65% vs 44% in the subset of patients with relapsed/refractory MZL, but with no difference in PFS between treatment arms in the relapsed/refractory MZL subgroup.35
In addition to higher ORRs, chemotherapy-naive patients treated with prior RTX had a more rapid time to initial response with ibrutinib than those treated with prior RTX-CIT, longer DOR, and longer PFS, although PFS and OS rates at 33 months did not differ according to prior therapy. Collectively, these results suggest that use of single-agent ibrutinib in earlier lines of therapy may optimize efficacy outcomes in patients with relapsed/refractory MZL.
With up to 45 months of ibrutinib treatment, the safety profile of single-agent ibrutinib remained consistent with observations during the primary analysis of this study and with published data on ibrutinib in previously treated NHL and chronic lymphocytic leukemia.23,36,37 With an additional 14 months of follow-up since the primary analysis, no new safety signals were observed. TEAEs were consistent in patients regardless of prior line of therapy and generally remained stable or decreased over time. Consistent with published data on ibrutinib that demonstrate a progressive decline in infections over time, the rate of grade 3 or higher infections was most frequent during the first year of ibrutinib treatment (n = 10), with few events occurring after the first year (n = 4).37-41 Of 5 patients with atrial fibrillation, three had events within the first year of treatment, consistent with previous reports that atrial fibrillation typically occurs within the first year of ibrutinib treatment and remains constant or declines over time.39-46 Atrial fibrillation was manageable without dose modifications or discontinuation of ibrutinib.
Previous MZL studies have identified NF-κB and Notch as 2 major signaling pathways that are disrupted through genetic alterations in key pathway genes.47 In this analysis, mutation of A20, a known negative regulator gene in the NF-κB pathway, was shown to be correlated to clinical efficacy (increased tumor shrinkage). Activating mutations in MYD88, a key signal transducer between toll-like receptor signaling and downstream NF-κB activation, were also positively associated with improved clinical efficacy (longer PFS). Mutations in CARD11 and KMT2D were associated with shorter DOR and worse PFS, whereas mutations in the Notch signaling pathway did not demonstrate any clinical impact. Although all correlations had P values <.05, these observations are nonetheless limited by the exploratory nature of the analysis and the small sample size, which was not powered for statistical significance. Taken together, the biomarker data suggest that ibrutinib’s clinical efficacy in MZL may be related to its disruption of the NF-κB signaling. Future studies in larger numbers of patients are warranted before treatment recommendations can be made on the basis of baseline genetics.
Conclusion
This final analysis of PCYC-1121 demonstrates the long-term safety and efficacy of treatment with single-agent ibrutinib in patients with relapsed/refractory MZL. Single-agent ibrutinib yielded responses regardless of prior line of therapy, although patients who had previous RTX treatment generally responded faster and better than those with prior RTX-CIT. Responses were consistent across MZL subtypes and deepened over time. These results support treatment with single-agent ibrutinib as an alternative to chemotherapy in this patient population with a favorable benefit-risk profile and convenient once-daily oral administration.
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Acknowledgments
The authors thank the patients for participating in the PCYC-1121 phase 2 study and their families.
This study was sponsored by Pharmacyclics LLC, an AbbVie Company. Medical writing support was provided by Melanie Sweetlove and funded by Pharmacyclics LLC, an AbbVie Company. C.R.F. is a CPRIT Scholar supported by a Cancer Prevention and Research in Texas grant to The University of Texas MD Anderson Cancer Center.
Authorship
Contribution: A.N., B.H., and I.A.-H. designed the study; A.N., S.d.V., M.C., P.M., C.R.F., F.M., G.P.C., S.M., C.T., S.P., S.D.S., J.C.B., and R.C. contributed to data collection; L.W.-K.C., S.W., and E.C. performed the data analyses; K.K., L.W.-K.C., S.W., E.C., and I.A.-H. confirmed the accuracy of the data and compiled it for analysis; all authors had access to the data and were involved in the interpretation of data, contributed to the manuscript review and revisions, and approved the final version for submission.
Conflict-of-interest disclosure: A.N. reports employment with Memorial Sloan Kettering Cancer Center; has received honoraria from Janssen, Medscape, Prime Oncology, and Pharmacyclics LLC, an AbbVie Company; has a consulting role with MorphoSys and Janssen; has received research funding from Rafael and Pharmacyclics LLC, an AbbVie Company; reports patent pending at Rafael; and has received travel expenses from Janssen and Pharmacyclics LLC, an AbbVie Company. S.d.V. has a consulting role with Bayer and Verastem. M.C. has stock ownership in Immunomedics; a consulting role with Pharmacyclics LLC, an AbbVie Company; has received research funding from AbbVie, Celgene, Genentech, BeiGene, and Pharmacyclics LLC, an AbbVie Company; and has received speaker fees from Celgene, Janssen, and Gilead. P.M. has consulting roles with AstraZeneca, Bayer, BeiGene, Cellectar, Celgene, I-MAB, Janssen, Karyopharm, Kite, MorphoSys, Sandoz, and TeneoBio; has received research funding from Karyopharm; and has received travel expenses from Janssen, MorphoSys and Bayer. C.R.F. has consulting roles with AbbVie, Spectrum, Celgene, Denovo Biopharma, OptumRx, Karyopharm, Pharmacyclics LLC, an AbbVie Company, Janssen, Gilead, and Bayer; has received research funding from AbbVie, Acerta, Celgene, Gilead, Genentech/Roche, Janssen, Millennium/Takeda, Pharmacyclics LLC, an AbbVie Company, and TG Therapeutics; and has received travel expenses from Genentech/Roche. C.T. has received honoraria from Gilead and Novartis; has consulting roles with Roche, Janssen, Celgene, Gilead, Kite, Novartis, Bayer, and Cellectis; has received research funding from Roche and Hospira; and has received travel expenses from Novartis, Roche, Janssen, Celgene, Novartis, and Cellectis. F.M. has received honoraria from Celgene, Roche, Bristol-Myers Squibb, and Gilead; and has consulting roles with Celgene, Roche, Epizyme, Gilead, and Bayer. G.P.C. has received honoraria from Takeda, Roche, Gilead, Bristol Myers Squibb, Merck, Celleron, ADC Therapeutics, Novartis, and Pfizer; has consulting roles with Takeda, Roche, Gilead, Bristol Myers Squibb, Merck, Celleron, and ADC Therapeutics; has received research funding from Bristol Myers Squibb, Celleron, Merck, Amgen, and Celgene; has received speaker fees from Takeda, Roche, Gilead, Novartis, and Bristol Myers Squibb; and has received travel expenses from Takeda and Roche; and has received support from NIHR Oxford Biomedical Research Centre. S.M. has consulting roles with AbbVie, AstraZeneca, Bioverativ, Genentech, Gilead, Janssen, Kite, Verastem, and Pharmacyclics LLC, an AbbVie Company; has received research funding from AbbVie, AstraZeneca, BeiGene, Janssen, Juno, Novartis, TG Therapeutics, and Pharmacyclics LLC, an AbbVie Company; and has received speaker fees from AstraZeneca, BeiGene, Janssen, and Pharmacyclics LLC, an AbbVie Company. S.P. has received research funding from AbbVie, Acerta Pharma, Amgen, BeiGene, Bristol Myers Squibb, Celgene, CTI Biopharma, Epizyme, Genentech, Gilead, GlaxoSmithKline, Janssen, Karyopharm, Merck, Rhizen, Seattle Genetics, TG Therapeutics, Verastem, and Pharmacyclics, an AbbVie Company; and has received speaker fees and travel expenses from Takeda, Celgene, and Johnson & Johnson. S.D.S. has consulting roles with AstraZeneca, Millennium/Takeda, and BeiGene; has received research funding from Acerta Pharma BV, AstraZeneca, Bayer, BeiGene, Denovo Biopharma, Genentech, Incyte Corporation, Merck, Portola Pharmaceuticals, Seattle Genetics, and Pharmacyclics LLC, an AbbVie Company; and reports spouse research funding from Ayala, Bristol Myers Squibb, and Ignyta. J.C.B. has received honoraria from Janssen; has consulting roles with Genentech, Gilead, Bayer, AstraZeneca, and Sandoz; and has received research funding from Oncternal Therapeutics. E.C. reports employment with Pharmacyclics LLC, an AbbVie Company, and stock ownership in AbbVie. S.W. reports employment, patents, royalties, and intellectual property with Pharmacyclics LLC, an AbbVie Company, and stock ownership in AbbVie. L.W.-K.C. reports employment, patents, royalties, and intellectual property with Pharmacyclics LLC, an AbbVie Company, and stock ownership in AbbVie and Eli Lilly. K.K. reports employment with Pharmacyclics LLC, an AbbVie Company, and stock ownership in AbbVie and Gilead. B.H. reports employment with Pharmacyclics LLC, an AbbVie Company, and stock ownership in AbbVie. I.A.-H. reports employment with Pharmacyclics LLC, an AbbVie Company, and stock ownership in AbbVie and Bristol Meyers Squibb. R.C. declares no competing financial interests.
Correspondence: Ariela Noy, Memorial Sloan Kettering Cancer Center, Hematology Division, Lymphoma Service, 1275 York Ave, New York, NY 10065; e-mail: noya@mskcc.org.