Key Points
Virus-specific T cells can be effectively used to treat BKPyV infections in HSCT and solid organ transplant recipients.
Virus-specific T cells against BKPyV are safe with no infusional toxicity, de novo graft-versus-host disease, or graft rejection.

Abstract
BK polyomavirus (BKPyV) infection is a major complication of hematopoietic stem cell transplant (HSCT) and solid organ transplant (SOT). Treatment options are limited, poorly effective, and have significant toxicities. Cellular therapy using T cells directed against BKPyV is an emerging therapy, and we report efficacy in controlling BKPyV-associated disease in highly immunocompromised patients. Virus-specific T cells (VSTs) against BKPyV were manufactured using either blood from the patient’s stem cell donor (donor-derived VSTs) or from unrelated donors (third-party VSTs). VSTs were used to treat BKPyV in 38 HSCT recipients and 3 SOT recipients between June 2017 and December 2019. Overall response rate was 86% in patients treated for BK viremia, 100% in patients treated for hemorrhagic cystitis, and 87% in patients treated for both BK viremia and hemorrhagic cystitis. No infusional toxicity, de novo graft-versus-host disease, or rejection of the organ occurred attributable to the VST infusion. BKPyV-specific immune responses were demonstrated by interferon-γ production by peripheral blood mononuclear cells postinfusion in response to BKPyV antigens. VSTs are a safe and potentially effective strategy to treat BKPyV and associated symptoms in recipients of HSCT and SOT. Cellular therapy should be considered for all patients with BKPyV and underlying immune suppression at risk of complications. This trial was registered at www.clinicaltrials.gov as #NCT02532452.
Introduction
Viral infections after hematopoietic stem cell transplant (HSCT) and/or solid organ transplant (SOT) are a leading cause of morbidity and mortality. BK polyomavirus (BKPyV), a member of the polyomavirus family, is particularly difficult to treat because of limited understanding of immunogenicity responses, lack of proven antiviral drugs, the appreciable side effects of available pharmacotherapy, as well as the need for prolonged hospitalization to manage the side effects of infection and treatment.
BKPyV has tropism for uroepithelium and can concentrate in the urine and induce p53-dependent apoptosis of urothelial cells, is associated with severe nephropathy after kidney transplantation, and is implicated as a causative pathogen in hemorrhagic cystitis after HSCT.1,2 Symptomatic hemorrhagic cystitis occurs in 25% of patients with BK viremia after HSCT, leading to morbidity, urinary obstruction, and possibly increased mortality.3-5
BKPyV nephropathy occurs in 5% of kidney transplant recipients, leading to chronic kidney disease (CKD), and can result in graft loss.6 BK viremia ≥10 000 copies/mL is classified as presumptive nephropathy after kidney transplant, even without renal biopsy. Recent studies also indicate that high-level BKPyV replication in HSCT recipients (≥10 000 copies/mL) is likely to result in kidney disease.3-5
A recent prospective BKPyV natural history study in pediatric HSCT recipients, reported by Laskin et al from 2 large pediatric institutions Cincinnati Children’s Hospital Medical Center (CCHMC) and Children’s Hospital of Philadelphia, identified a high incidence (54%) of BKPyV viremia and demonstrated that high levels of viremia (>10 000 copies/mL), whether symptomatic or not, were associated with a severe reduction in kidney function and a need for dialysis.7 CKD markedly increases the risk of cardiovascular disease because of accelerated atherosclerosis caused by endothelial damage and concomitant hypertension, indicating the importance of avoiding this complication.8-10 Furthermore, patients who undergo HSCT and progress to CKD are 16 times more likely to develop end-stage renal disease, and those needing chronic dialysis have mortality rates of 90%, much higher than other patients with end-stage renal disease.11,12 In addition, BKPyV viremia is associated with a higher risk of developing thrombotic microangiopathy, graft-versus-host disease (GVHD), and death.
Cidofovir, leflunomide, and administration of immunoglobulins to HSCT recipients are commonly prescribed to treat BKPyV in the absence of other proven therapeutic options with limited efficacy; thus, the current mainstay of treatment is supportive care. However, the prospective cohort study showed that children who received cidofovir were no more likely to clear BKPyV than those who did not, but did have a significant reduction in their glomerular filtration rate, suggesting this treatment should be avoided. The cohort study did show that recovery of endogenous BKPyV-specific T cells (VSTs) was associated with viral clearance, similar to prior studies after kidney transplantation, suggesting that VSTs could be an effective approach to eliminate BKPyV in immunocompromised recipients.7
Multiple studies have demonstrated that VST products with specificity for commonly detected viruses (eg, Epstein-Barr virus [EBV], cytomegalovirus [CMV], adenovirus) can be safely administered to pediatric transplantation recipients receiving allogeneic HSCT without inducing GVHD.13-17 Advances in the production of trivalent VSTs have dramatically reduced production time to 2 to 3 weeks by stimulation of T cells with antigen-presenting cells pulsed with overlapping peptide pools spanning entire protein sequences of target viral antigens.
Here, we present our experience manufacturing BKPyV-VSTs and treating patients with BKPyV after HSCT or SOT. In a phase 2 clinical trial, we now show that administration of BKPyV-specific T cells derived from a patient’s HSCT donor or alternatively, a third-party donor, can reduce symptomatic infection and BK viral load in HSCT and solid organ transplant recipients.
Methods
Study population and clinical trial
This phase 2 study included HSCT and SOT recipients who developed BKPyV viremia and/or hemorrhagic cystitis or nephropathy after transplant (a subset of the larger cohort who received VSTs for other viral infections). The study was approved by the CCHMC institutional review board and the US Food and Drug Administration. HSCT and SOT recipients were consented to receive transplant donor-derived and/or third-party BKPyV VSTs after meeting study eligibility criteria.
The primary study end point was successful production of healthy donor-derived BKPyV-VSTs and infusion into HSCT and SOT recipients without toxicity. The secondary end point was antiviral efficacy of the infused BKPyV-VSTs.
PBMC collection
Quadrivalent VSTs specific for EBV-, adenovirus-, CMV-, and BKPyV-specific cytotoxic T lymphocytes (VSTs) were manufactured using peripheral blood mononuclear cells (PBMCs) from 2 sources: stem cell donor-derived PBMCs and unrelated donor (third-party) PBMCs. All donors underwent required screening procedures within 7 days of PBMC collection. All unrelated donors for third-party VST infusions met US Food and Drug Administration criteria for donor eligibility.
Donor-derived VSTs
Study enrollment for manufacture of donor-derived VST was offered to transplant recipients and their respective stem cell donors (>10 kg weight) when the risk of viral reactivation after transplant was deemed to be high, defined as recipients of: (1) grafts from unrelated donors, (2) T-cell depleted grafts, (3) grafts from family members mismatched at 1 or more HLA-A, HLA-B, HLA-C, or HLA-DRB1 locus, and (4) transplants that include in vivo T-cell depletion with alemtuzumab or antithymocyte globulin (ATG).
HSCT recipients and their respective stem cell donors were consented for study participation. Related donors were consented by study staff and blood was collected on site. The protocol was also approved by the National Marrow Donor Program institutional review board, and unrelated donors were consented for donation of additional blood (up to 80 mL) for VST generation by the National Marrow Donor Program donor center and were shipped to CCHMC. VSTs were cryopreserved until a recipient met the eligibility criteria for VST infusion.
Third-party VSTs
PBMCs were obtained from consented adult volunteers (eg, platelet donors) by study staff, specifically for VST generation for third-party use. Additional donors with HLA types of interest who had already been HLA typed for other reasons were recruited via scheduled blood donor drives. In addition, donor-derived VSTs were transferred from the donor-derived to the third-party donor VST bank when products were no longer needed for patient specific use.
VST manufacturing process
VST manufacturing methods were modified from reported strategies.17 In brief, PBMCs were separated from whole blood by density centrifugation (Ficoll-Paque Premium, GE Healthcare). Either fresh or frozen/thawed PBMCs were then pelleted and stimulated with pepmixes that span the antigenic epitopes of adenovirus (Hexon-AdV3 and Hexon-AdV5), CMV (pp65 and IE1), EBV (EBNA1, LMP2, and BZLF1), and BKPyV (VP1 and large T) at 10 ng/1 × 106 cells for 30 minutes at 37°C. The cells were then suspended in a culture media consisting of 45% Advanced RPMI1640, 45% Click medium, 10% fetal bovine serum, 400 U/mL interleukin-4, and 10 ng/mL interleukin-7 and cultured for 11 to 12 days in G-Rex 10M culture devices at 37°C and 5% CO2. Cultures were maintained by replenishing with cytokines on day 3 or 4 then harvesting and cryopreserving the VST products on day 11 or 12. All products were tested for viability, sterility, alloreactivity, and T-cell phenotype, and only infused if all release criteria were met (supplemental Table 1). VST products were characterized by flow cytometric analysis of interferon-γ (IFN-γ) production in response to repeat stimulation with BKPyV pepmixes (supplemental Appendix 1). All patients enrolled had a suitable VST product available for clinical use.
ELISPOT assays
VST specificity to individual viruses and expansion of virus specific T cells in the recipient were measured by the interferon γ enzyme-linked immunospot (ELISPOT) assay. The ELISPOT assay was performed to quantitate the number of T cells in the peripheral blood of VST recipients secreting IFN- γ when stimulated with pepmixes of BKPyV viral proteins VP-1 and LTA (JPT Peptide Technologies, Berlin, Germany). The assay was performed using the Human IFN-γ ELISPOTPLUS kit (Mabtech, Nacka Strand, Sweden). Briefly, PBMCs were collected from recipients before VST infusion and weekly for at least the first 4 weeks after infusion. PBMCs were incubated with 0.5 µg/mL of BKPyV pepmixes for 14 to 16 hours. Spot forming cells (sfc) were developed according to manufacturer recommendations and quantitated using the Immunospot S6 analyzer (Cellular Technology Limited, Cleveland, OH).
VST selection and release
For HSCT recipients, donor-derived VSTs, if available, were preferred over third-party VSTs for the first infusion after meeting infusion criteria because the donor-derived product was deemed to have less associated risk than third-party VST product in our initial studies. Subjects with persistent BKPyV illness who received a donor-derived VST product without a clinical response could receive third-party VSTs 1 month after donor-derived VST infusion. Third-party donor VSTs may be cleared from circulation within a period of weeks because of high levels of HLA mismatch, so serial VST infusions (with the same or different units) were allowed at monthly intervals if the study subject was deemed to have clinical benefit from a prior VST infusion, but had not completely achieved desired clinical response and did not have documented acute GVHD within 30 days from previous infusion. Additionally, multiple VST infusions were allowed in single patients if they had new episodes of viremia or cystitis following the initial post-VST monitoring period.
For subjects receiving third-party VSTs, the most closely HLA-matched VST unit (ideally matched at least 1 class I and 1 class II allele) was selected. If there were equivalent HLA-matched VST units, the unit with higher BKPyV activity was selected. VST release criteria and characteristics are listed in supplemental Table 1.
VST infusion and monitoring
VSTs were infused into eligible and consented HSCT or SOT recipients who had documented BKPyV reactivation or BKPyV-associated complications defined as any of the following: (1) plasma BKPyV polymerase chain reaction (PCR) >1000 copies/mL or (2) evidence of symptomatic BKPyV infection, which may include symptomatic hemorrhagic cystitis (grade ≥2) or BKPyV nephropathy. HSCT recipients were required to be at least 28 days after stem cell transplant and must have achieved neutrophil engraftment as documented by absolute neutrophil count recovery ≥500 × 103/μL postnadir. Clinical status must allow tapering of steroids to ≤0.5 mg/kg prednisone equivalent.
Exclusion criteria for VST infusion included active acute GVHD grades II-IV (in HSCT recipients), uncontrolled bacterial or fungal infection, uncontrolled relapse of malignancy, and infusion of ATG or alemtuzumab within 2 weeks of VST infusion.
Study subjects received a fixed cell dose of up to 5 × 107 VSTs/m2 infused intravenously using a standard operating procedure for infusion of therapeutic T cells. Patients could receive up to 12 infusions repeated monthly if needed, provided they did not have any adverse effects from their previous VST infusion including the development of GVHD. There was no dose escalation.
Patients were not prescribed any additional antiviral treatments after the VST infusion, unless there was an evidence of progression of invasive viral disease or greater than a 1-log increase in viral titer more than 2 weeks after VST infusion, or additional antiviral therapy was deemed clinically necessary.
Clinical and cellular response criteria
Viral load was monitored by quantitative PCR at weekly intervals for a minimum of 4 weeks post-VST infusion. BKPyV response was evaluated 4 weeks after last VST infusion and/or 4 weeks after each VST infusion in subjects who received multiple VST infusions. Clinical response to VST therapy was assessed by the principal investigator and designees based on reduction in hemorrhagic cystitis symptomatology and required medical interventions, reduced need for blood product support, and discontinuation of urethral catheters. Complete response (CR) was defined as undetectable plasma BKPyV PCR within the 4-week period after last VST infusion, or resolution of hemorrhagic cystitis below grade 2 and no clinical cystitis symptomatology requiring medical therapy (analgesic medications). Partial response (PR) was defined as >50% reduction in plasma BKPyV PCR quantification within the 4-week period following VST infusion, or reduction of hemorrhagic cystitis by at least 1 grade and associated clinical symptoms. No response was defined as either no change or an increase in BKPyV PCR number within the 4-week period following VST infusion, or no change to hemorrhagic cystitis symptomatology.
ELISPOT assays were performed at weekly intervals for a minimum of 4 weeks post-VST infusion to determine the presence of BKPyV-specific T cells in the circulation. Background sfc counts in media were subtracted from observed counts to determine absolute sfc counts.
Results
HSCT recipients’ disease characteristics and therapy response
A total of 38 HSCT recipients received BKPyV VSTs; 20 were treated for BK viremia, 4 for hemorrhagic cystitis, and 14 for both BK viremia and hemorrhagic cystitis between June 2017 and December 2019.
Demographics of HSCT patients are shown in Table 1. BKPyV viremia was detected at a median of 28 days (range, 5-187 days) after transplant. Hemorrhagic cystitis was diagnosed in 18 patients a median of 20.5 days (range, 2-121 days) after HSCT. Patients had grade 2 (n = 10), 3 (n = 6), or 4 (n = 2) hemorrhagic cystitis requiring medical care, including narcotic medications for pain management. Ten patients had additional viral reactivations/infections as well as BKPyV and were receiving specific antiviral medical therapy for those viremias (Table 2). The median absolute lymphocyte count at the time of VST infusion was 630 cells/μL with a range of 0 to 13 500.
Fourteen of the 38 HSCT patients received at least 1 infusion with a donor-derived VST product, whereas 28 patients received third-party VSTs. Four patients received donor-derived followed by third-party VST products. Median BKPyV viral load before first VST infusion was 2100 copies/mL (range, 135-1440 951 copies/mL) in donor-derived VST recipients and 12 543 copies/mL (range, 1000-5 513 000 copies/mL) in third-party VST recipients. The median time from eligibility for VST infusion and VST infusion was 5 days. The median number of VST infusions administered was 1 (range, 1-3) for donor-derived VST recipients and 1 (range, 1-10) for third-party VST recipients. There were no documented infusion reactions. No patient experienced de novo GVHD during the study period (4 weeks after infusion). One subject with a prior history of acute skin GVHD that had resolved before VST infusion developed acute stage 2 skin GVHD 5 days after receiving their first infusion of third-party donor VSTs. The GVHD resolved completely after systemic steroid therapy; this patient did not receive any additional VSTs and his BKPyV and cystitis resolved after 3 months.
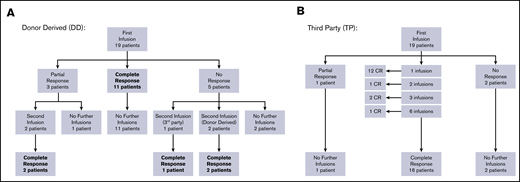
Overall response per infusion was 86% for subjects with BKPyV viremia demonstrating response to therapy with 69% achieving a CR (clearing viremia) and 17% achieving PR (>50% reduction in viremia). All study subjects with hemorrhagic cystitis responded to VST therapy with 75% achieving CR and 25% PR. In the subgroup of patients who had both viremia and hemorrhagic cystitis, antiviral response was documented in 87% of infusions (58% CR and 29% PR). Additional details of response based on VST type received are displayed in Figures 1A-B and Table 2. Results of the 10 patients who had concomitant viral infections at the time VST infusion are described in supplemental Table 2.
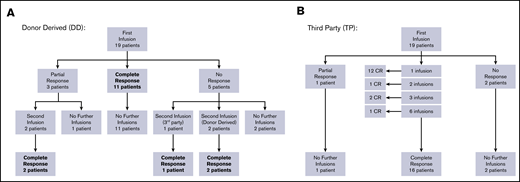
Response to VSTs. (A) Response to donor-derived BKPyV-VST flowchart. (B) Response to third-party BK-VST flowchart.
Response to VSTs. (A) Response to donor-derived BKPyV-VST flowchart. (B) Response to third-party BK-VST flowchart.
Patients who received donor-derived VSTs for BKPyV viremia (N = 6) showed an overall response rate per infusion of 75%, whereas those receiving third-party VSTs (N = 14) had an overall response rate of 95% (Table 2). In patients with hemorrhagic cystitis alone, those receiving third-party VSTs (N = 2) both achieved CR, whereas in patients receiving donor-derived VST (N = 2), 1 achieved CR and 1 PR. These data taken together indicate good response and low toxicity with third-party VSTs.
SOT recipients BKPyV viremia and nephropathy are responsive to VSTs
Three SOT recipients were included in this study (Table 3): 1 had received a heart transplant; 1 patient was a kidney transplant recipient; and 1 patient had both kidney and heart transplants. All patients received third-party “off-the-shelf” VSTs and received graft rejection prophylaxis with tacrolimus/everolimus and low-dose prednisone at the time of VST treatment. One patient had a CR, whereas the other 2 patients had a PR, including 1 patient who had concurrent resolution of EBV-posttransplant lymphoproliferative disorder (PTLD). No patients experienced toxicity or graft rejection.
BKPyV-specific T cells expanded in vivo post-VST infusion
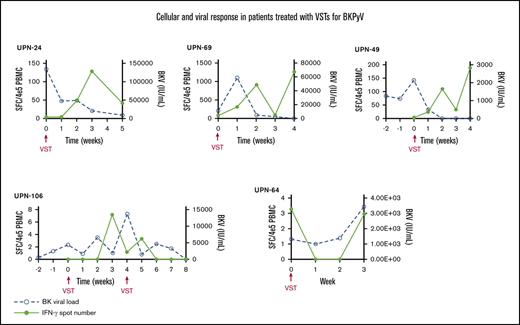
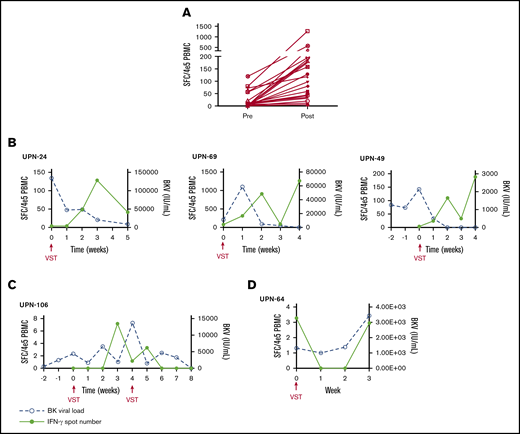
PBMCs were collected at the time of infusion and then weekly for the first month after each independent infusion to detect expansion of BKPyV-specific T cells by interferon-γ ELISPOT. Adequate samples were available to perform ELISPOT on 33/38 patients, including 11 patients who received donor-derived VST infusions, 21 patients who received third-party VST infusions, and 1 patient who received both donor-derived followed by third-party VST. ELISPOT data were available from all 3 SOT recipients. ELISPOT assay was performed on 30 patients who ultimately achieved a clinical response; 23/29 (79.3%) of these patients had an increase in BKPyV-specific T cells detected in the peripheral blood after the first infusion at the time when clinical responses were achieved. The median number of BKPyV-specific T cells increased from 4.8 sfc/4 × 105 (range, 0-120.5) to a median peak of 79.8 sfc/4 × 105 (range, 3.7-1265.0) (Figure 2A). Representative examples are shown for patient where only 1 infusion was required and when more than 1 infusion was needed to maintain good control of viral disease (Figure 2B-C). In the 4 patients who never achieved any response from VST therapy, ELISPOT accordingly showed no increase in BKPyV-specific T-cell frequency, as shown with a representative patient in Figure 2D.
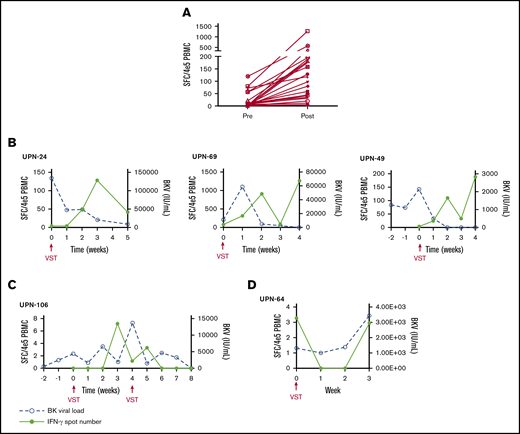
Characteristics of the cellular and viral response. (A) The median number of BKPyV-specific T cells pre- and post-VST infusion. Interferon-γ response and corresponding viral PCR in patients who require 1 VST infusion (B), in a patient requiring multiple VST infusions (C), and in 1 patient who did not respond to VST infusion (D).
Characteristics of the cellular and viral response. (A) The median number of BKPyV-specific T cells pre- and post-VST infusion. Interferon-γ response and corresponding viral PCR in patients who require 1 VST infusion (B), in a patient requiring multiple VST infusions (C), and in 1 patient who did not respond to VST infusion (D).
Discussion
We report our findings using VSTs generated ex vivo to treat BKPyV viremia and/or cystitis in patients after HSCT or SOT. The data show encouraging efficacy, with a majority of those treated having a clinical antiviral response. Administration of VSTs was associated with minimal toxicity. No infusion reactions were recorded and the incidence of new or worsening GVHD was low and resolved with anti-GVHD treatment. Several prior studies have reported activity of VSTs to control virus infections such as CMV, EBV, and adenovirus.13,15,17
Tzannou et al also reported the use of third-party VSTs to treat BKPyV in 14 patients with hemorrhagic cystitis and 2 with BKPyV nephropathy.14 These authors reported symptomatic and virologic response in all cases, in agreement with our finding of a high response rate in both resolution of hemorrhagic cystitis and of viremia. Similarly, Papadopoulou et al demonstrated clinical responses of VSTs to treat BKPyV and hemorrhagic cystitis and showed efficacy in treating multiple concurrent viral infections with a single VST product.17 We believe that our study is the first focused exclusively on BKPyV and documents the largest number of BKPyV cases treated with VST products to date. We also report the first experience using VSTs to treat solid organ transplant recipients infected with BKPyV.
BKPyV-driven hemorrhagic cystitis and viremia are important complications of HSCT, associated with high morbidity and resource use, as well as increased mortality. Our group recently reported a prospective 2-center study of BKPyV viremia and viruria in 193 children and young adults undergoing allogeneic HSCT.7 Eighteen percent of the participants in this epidemiological study had viremia >10 000 copies/mL and 45% viruria >109 copies/mL in the first 3 months after HSCT. Importantly, among the 147 patients without cystitis, in whom BKPyV viremia would likely not routinely be tested clinically, 40% had some degree of viremia, and in one-third of these cases viremia was >10 000 copies/mL. These data raise the question as to whether BKPyV viremia without symptomatic cystitis is of clinical significance. We discovered that glomerular filtration rate was significantly reduced 2 years after HSCT in asymptomatic cases with BKPyV viremia >10 000 copies/mL compared with those without viremia. Moreover, BKPyV viremia of >10 000 copies/mL was associated with a sevenfold increased risk of transplant-associated thrombotic microangiopathy. These findings suggest there might be value in routine screening for BKPyV viremia in HSCT recipients so treatment could be initiated to reduce these adverse effects. Treatment of BKPyV has previously been limited, and cidofovir is the only antiviral agent in common use. The prospective cohort study identified that use of cidofovir was not associated with clearance of BKPyV viremia; in fact, BKPyV was less likely to clear in those treated with cidofovir. These data are similar to findings reported in a retrospective study of BKPyV hemorrhagic cystitis reported from Seattle,18 though our onset of hemorrhagic cystitis is earlier than previously reported, which may reflect our screening practices and cohort of patients. Importantly, we did show that clearance of BKPyV was associated with recovery of BKPyV-specific T cells in the host, supporting our current strategy of infusing ex vivo-generated VSTs.
In our study, we report outcomes of both donor-derived (patient-specific) and third-party (off-the-shelf) VST infusions. Our data suggest that outcomes are similar using either product, an important observation, in agreement with reports of others, as dissemination of VST technology to multiple sites is easier without the need for patient-specific product.13,19,20 The key expected benefit of donor-derived VSTs is long-term persistence of the T cells in the circulation because the product is HLA-matched with the host. Prior reports suggest that donor-derived VSTs can be detected as long as 10 years after infusion.21 Prior data have also demonstrated shorter persistence of third-party VSTs; this would be expected because third-party VSTs are significantly mismatched with the recipient and can be rejected by endogenous T cells.22 A small number of patients required multiple infusions of VSTs to control disease, but the majority did not see recurrence of BKPyV viremia/disease after achieving a CR, suggesting that the third-party VSTs persisted until endogenous immune reconstitution, and/or suggesting that low levels of persistence were sufficient to control viral disease.
The use of third-party, or off-the-shelf VSTs is logistically attractive because of their immediate availability. We showed effectiveness of these products, despite minimal HLA matching between VSTs and recipient. Additionally, we did not observe clinically concerning graft failure/rejection or GVHD, which is a potential complication of using HLA mismatched T cells in the SOT or HSCT settings. The minimal evidence of alloreactivity and complications of GVHD and graft rejection is likely because of terminally differentiated characteristics of the T cells in the product, which comprise CD4 and CD8 effector T cells, with few naïve T cells present.
Our study included 3 recipients of solid organ transplants with BKPyV viremia. One of these patients also had EBV-driven PTLD. The usual approach to manage BKPyV viremia in solid organ transplant recipients is reduction in immune suppression to allow control of virus by the host immune system. This strategy can be successful, but in a proportion of cases, viremia may persist, and over time may lead to reduced renal function and active nephropathy. Moreover, although immune suppression can be reduced in renal transplant recipients, this is a riskier strategy in recipients of heart, lung, and liver transplants. The absence of effective antiviral strategies led us to test VSTs in this setting, without additional modification of immune suppression. Our preliminary results in this small group of cases show that these patients received clinical benefit without adverse effects, indicating that these T cells can expand in the presence of immunosuppressive medications without evidence of rejection and need for reduction of immunosuppression, supporting further investigation of this strategy, which is ongoing.
We demonstrated a high response rate of BKPyV to VSTs in this study, but not all cases had an optimal response. It is important to understand the characteristics of effective vs ineffective VST products to optimize manufacture and selection of the best third-party units in the future. One reason for poor performance is the lack of prior donor exposure. In addition, several of our donor-derived units demonstrated no in vitro BKPyV-specific activity by flow cytometry. In this study, we elected to infuse those VST products and did observe responses in some (n = 4/18; 22%). However, now that we are confident regarding the safety and efficacy of third-party VSTs, we would choose a third-party VST product as first choice in this setting. A second possible cause of reduced efficacy is limited VST persistence in vivo with the mismatched third-party VST infusions. This was a particular concern in the recipients of SOT, who may be more immune competent than HSCT recipients early after transplant; in 1 of the SOT recipients, monthly third-party infusions were administered safely to maintain antiviral activity in vivo. Interestingly, the 3 recipients of VSTs who had prior SOT all showed minimal expansion of BKPyV-specific T cells in vivo by ELISPOT assay despite achieving an objective clinical response. This phenomenon could be due to rapid host rejection by comparatively immunocompetent individuals as compared with HSCT recipients, although this did not seem to affect efficacy, as demonstrated by important work from Prockop et al that showed reduced numbers of circulating VSTs in SOT recipients compared with HSCT recipients treated for EBV-associated lymphoma.23 A third reason for failure of VSTs could be poor antigen presentation, perhaps because of the VST product recognizing viral antigen through HLA alleles that are not shared with the recipient. There is emerging evidence of what HLA-restricted epitopes are important in controlling BKPyV infection,24 though further work is needed to identify key BKPyV HLA-restricted epitope-specific responses, and these data will inform future selection of third-party VST products for infusion. It is important to note however that our study design allowed infusion of a different VST product if the first VST infusion failed to clear BKPyV, and in this study we did observe antiviral responses after these additional infusions.
Postinfusion ELISPOT data were available for 89% of the patients in this cohort, demonstrating this assay is a feasible way to demonstrate expansion of BKPyV-specific T cells in patient peripheral blood after VST infusion. Interestingly, almost 20% of patients who had a clinical response to VSTs did not show any increase in BKPyV T-cell frequency by ELISPOT. Four of 6 of these patients had hemorrhagic cystitis at infusion, with an additional patient developing cystitis shortly after his first infusion. Therefore, one possibility is that VSTs preferentially trafficked to urothelial tissue rather than the peripheral blood in these settings. Second, additional epitope spreading (through VP2 and/or VP3) could explain the observed clinical response that was not able to be tracked with ELISPOT monitoring. An important caveat with ELISPOT data is that we cannot definitively state that the detected BKPyV-specific T cells are derived from the infused product rather than endogenous host- or graft-derived T cells. However, the rapid kinetics of clinical improvement related to VST infusion and the relative lymphopenia of this cohort (median absolute lymphocyte count at each infusion was 630 cells/μL with a range of 0-13 500) would support the assertion that these T cells are derived from the VST product.
This study demonstrates anti-BKPyV responses in patients who received donor-derived and/or third-party VST infusions with minimal toxicity. These data are important because there are currently no effective drug treatments for BKPyV. The availability of an effective and safe treatment of BKPyV viremia means that consideration should be given to increased PCR screening for BKPyV, even in the absence of symptoms. The high frequency of viremia, including asymptomatic viremia, also raises the consideration of administration of BKPyV-directed VSTs to all HSCT recipients early after HSCT. In the context of a review article, Barrett et al reported prophylactic infusion of VSTs targeting BKPyV into 9 patients 14 days after HSCT and none developed hemorrhagic cystitis.25 Hence, we are currently testing prophylactic administration of VSTs to test this strategy to prevent viremia before the development of hemorrhagic cystitis. If effective, prophylactic BKPyV VSTs given routinely after HSCT could reduce early morbidity and late renal injury significantly.
Send data sharing requests via e-mail to the corresponding author, Adam S. Nelson, adam.nelson@cchmc.org.
Acknowledgments
The authors thank the staff at the Hoxworth Blood Center for their assistance with manufacturing VSTs and infusion capabilities, the care managers for helping coordinate patient care, and patients and their families for participating in this study.
S.J. received National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development funding (RO1 HD093773) for the study drug provided by Alexion Pharmaceuticals.
Authorship
Contribution: A.S.N., S.M.D., and M.S.G. designed the study, performed research, and wrote the paper; C.L., J.D.R., and X.Z. performed functional studies and analysis; D.H. and T.L. performed VST manufacturing; A.S., S.J., S.T., J.A.C., M.K., C.M.B., and P.J.H. provided vital conceptual insights for study design, assisted with study subject accrual and data collection, and prepared figures; and all authors reviewed and edited the manuscript.
Conflict-of-interest disclosure: C.M.B. is on the advisory board for Cellectis and is on the scientific advisory boards for Catamaran Bio and Mana Therapeutics with stock and/or ownership, is on the board of directors for Caballeta Bio with stock options, and has stock in Neximmune and Torque Therapeutics. P.J.H. is a cofounder and on the board of directors of Mana Therapeutics and has filed intellectual property related to virus-specific T cells. S.M.D. has a US pending patent application under review, received research support from Alexion Pharmaceuticals, and consultancy from Novartis. S.J. has a US pending patent application under review and received travel support and consultancy fees from Omeros (unrelated to this work). The remaining authors declare no competing financial interests.
Correspondence: Adam S. Nelson, Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229; e-mail: adam.nelson@cchmc.org.