Key Points
12-LOX oxylipins of DPA inhibit platelet activation and clot formation.
DPA oxylipins inhibit platelet function through a selective PPARα nongenomic mechanism.

Abstract
Arterial thrombosis is the underlying cause for a number of cardiovascular-related events. Although dietary supplementation that includes polyunsaturated fatty acids (PUFAs) has been proposed to elicit cardiovascular protection, a mechanism for antithrombotic protection has not been well established. The current study sought to investigate whether an omega-6 essential fatty acid, docosapentaenoic acid (DPAn-6), and its oxidized lipid metabolites (oxylipins) provide direct cardiovascular protection through inhibition of platelet reactivity. Human and mouse blood and isolated platelets were treated with DPAn-6 and its 12-lipoxygenase (12-LOX)–derived oxylipins, 11-hydroxy-docosapentaenoic acid and 14-hydroxy-docosapentaenoic acid, to assess their ability to inhibit platelet activation. Pharmacological and genetic approaches were used to elucidate a role for DPA and its oxylipins in preventing platelet activation. DPAn-6 was found to be significantly increased in platelets following fatty acid supplementation, and it potently inhibited platelet activation through its 12-LOX–derived oxylipins. The inhibitory effects were selectively reversed through inhibition of the nuclear receptor peroxisome proliferator activator receptor-α (PPARα). PPARα binding was confirmed using a PPARα transcription reporter assay, as well as PPARα−/− mice. These approaches confirmed that selectivity of platelet inhibition was due to effects of DPA oxylipins acting through PPARα. Mice administered DPAn-6 or its oxylipins exhibited reduced thrombus formation following vessel injury, which was prevented in PPARα−/− mice. Hence, the current study demonstrates that DPAn-6 and its oxylipins potently and effectively inhibit platelet activation and thrombosis following a vascular injury. Platelet function is regulated, in part, through an oxylipin-induced PPARα-dependent manner, suggesting that targeting PPARα may represent an alternative strategy to treat thrombotic-related diseases.
Introduction
Cardiovascular disease is the leading cause of death, and platelets play an essential role through regulation of arterial clot formation mediating the hemostatic and thrombotic responses to vascular insult.1 Recently, the REDUCE-IT and VITAL trials2-7 have shown that dietary supplementation of polyunsaturated fatty acids (PUFAs) or their synthetic analogs may significantly reduce cardiovascular-related diseases that are due to dyslipidemia and atherothrombotic complications. Although these studies show significant cardioprotective properties of PUFAs through decreasing triglycerides and protection from myocardial infarction, little is known about the mechanistic underpinnings by which PUFAs elicit protection in the blood. This lack of mechanistic insight reveals the need for new studies to assess how PUFAs prevent arteriothrombotic events through regulation of platelet activation.
To delineate which PUFAs and their bioactive oxidized lipids (oxylipins) exhibit demonstrable antiplatelet properties, we screened omega-3 and omega-6 PUFAs for their ability to regulate platelet function.8 This work identified that omega-6 PUFAs, such as dihomo-γ-linolenic acid (DGLA) and its 12-lipoxygenase (12-LOX) oxylipin, 12(S)-hydroxyeicosatetraenoic acid (12(S)-HETrE), exert antithrombotic effects through the prostacyclin receptor in platelets.9,10 These studies provided insight on the efficacy of exogenous PUFAs, such as DGLA, in regulating platelet function.
In contrast to DGLA, a highly abundant, but previously unstudied fatty acid, docosapentaenoic acid (DPA), produces oxylipins through the omega-3 or omega-6 PUFA pathway in the blood. These oxylipins are derived from omega-3 all-cis-7Z,10Z,13Z,16Z,19Z-docosapentaenoic acid (DPAn-3) or omega-6 all-cis-4Z,7Z,10Z,13Z,16Z,19Z-docosapentaenoic acid (DPAn-6). DPAn-3 has been shown to be metabolized in a lipoxygenase-dependent manner to 11-hydroxy-docosapentaenoic acid (11-HpDPAn-3) or 14-HpDPAn-3 acid in human platelets.11 DPAn-6 has been demonstrated to be metabolized to 14-hydroxy-docosapentaenoic (14-HDPAn-6).12-14 Although 1 study reported that DPAn-3 inhibited collagen or U46619-mediated platelet aggregation by directly inhibiting cyclooxygenase-1 (COX-1) activity15 and by accelerating COX-1 isomer product formation, the current study supports that the antiplatelet effects of these PUFAs are primarily mediated through their oxylipins generated by 12-LOX, and not through regulation of COX-1.
The objective of this study was to elucidate the mechanism by which DPA and its oxylipins inhibit platelet reactivity. We show for the first time that DPAn-6 and its oxylipins, 11S-hydroperoxy-4Z,7Z,9E,13Z,16Z-docosapentaenoic acid (11-HpDPAn-6) and 14S-hydroperoxy-4Z,7Z,10Z,12E,16Z-docosapentaenoic acid (14-HpDPAn-6), potently inhibit platelet activation. DPAn-6 oxylipins did not induce G protein α s subunit (Gαs) signaling effectors, as has been shown previously for the DGLA oxylipin regulation of platelets.8-10 Instead, these oxylipins selectively mediated their inhibitory effect on platelet function through activation of the peroxisome proliferator activator receptor-α (PPARα), a member of the nuclear receptor superfamily. Further, platelets from PPARα-deficient mice (PPARα−/−) were unable to respond to DPAn-6–derived oxylipins compared with wild-type (WT) platelets in ex vivo and in vivo assays of platelet function and thrombosis. Altogether, this study demonstrates that targeting PPARα may be an attractive therapeutic strategy, especially in the context of thrombosis prevention.
Methods
Expression and purification of h12-LOX
Recombinant expression and purification of WT human platelet 12-lipoxygenase (h12-LOX) were performed as previously described.16 The metal content was assessed on a Finnigan inductively coupled plasma mass spectrometer.
Steady-state kinetics of h12-LOX
Reactions of h12-LOX were performed in 2 mL of 25 mM HEPES buffer (pH 8.0) at 25°C. PUFA concentrations ranged from 0 to 20 μM, and product formation was monitored by measuring absorbance at 234 nm using ε234 = 25 000 M/cm for conjugated diene formation in buffer. Reactions were initiated by the addition of h12-LOX and were monitored on a PerkinElmer Lambda 45 UV-Vis spectrophotometer.
Synthesis and purification of oxylipins
14-HpDPAn-6 and 11-HpDPAn-6 were synthesized using recombinant h12-LOX in 200 mL of 25 mM HEPES buffer at pH 8.0. The oxylipin products were purified using high-performance liquid chromatography with a normal phase phenomenex silica column (5 μm, 250 mm × 10 mm) and an isocratic mixture of 99% hexane, 1% isopropanol, and 0.1% trifluoroacetic acid. The products were identified using retention time, fragmentation patterns, and UV absorption; purity was checked by liquid chromatography with tandem mass spectrometry (LC-MS/MS).
Oxylipin product analysis from platelet and fatty acid reactions
Supernatants from washed platelets were extracted with dichloromethane, followed by the addition of molar excess of trimethyl phosphite, and then dried under nitrogen gas. The products were resuspended in 50/50 acetonitrile and 0.1% formic acid water, and 90 μL was injected for LC-MS/MS analysis.
Human blood collection and platelet preparation
Written informed consent was obtained from all volunteers under the approval of the University of Michigan Institutional Review Board prior to blood draws. Blood draws from male and female donors were collected into 3.2% sodium citrate–containing Vacutainers, and platelets were purified by centrifugation, as previously described.17 Platelets were resuspended in Tyrode’s buffer at 3 × 108 platelets per milliliter.
Mouse blood collection and washed platelet preparation
All experimental procedures in this study were approved by the University of Michigan Institutional Animal Care and Use Committee. WT (B6129SF2/J) and PPARα-knockout (PPARα−/−) mice were purchased from The Jackson Laboratory. Blood was collected from the inferior vena cava of 12- to 16-week-old anesthetized male and female mice. Platelets were resuspended in Tyrode’s buffer at 3 × 108 platelets per milliliter.
Dietary supplementation in mice
At 8 weeks, B6129SF2/J mice (male and female) were orally gavaged daily with vehicle control (polyethylene glycol 300), 50 mg/kg DGLA, or 50 mg/kg DPAn-6 for 4 weeks prior to blood collection and platelet preparation (Nu-Chek Prep, Inc). This is equivalent to a 3.5-g supplementation of PUFA in a 70-kg person.
Platelet aggregation
Washed platelets were treated with the selected PUFAs, oxylipins, or PPAR antagonists and agonists at the indicated time prior to platelet aggregation in a Lumi-Aggregometer (CHRONO-LOG Model 700D), under stirring conditions (1200 rpm) at 37°C.
Calcium mobilization
Human platelets (1 × 106) were treated with PUFAs or oxylipins in the presence of 0.5 μg of the calcium dye Fluo-4 AM (Thermo Fisher Scientific) for 10 minutes at 37°C and recalcified with 1 mM CaCl2. Fluorescence intensity of agonist-stimulated calcium flux was monitored in real-time by flow cytometry (BD Accuri C6).
Protein kinase C activation
Mouse platelets (3 × 108/mL) were treated with PUFA or oxylipins for 15 minutes prior to stimulation with thrombin or convulxin for 5 minutes and quenched in 5× sample buffer. Samples were immunoblotted for phospho-(Ser) protein kinase C (PKC) substrate (Cell Signaling Technology) and pleckstrin (BD Biosciences) antibodies.
Dense granule secretion
Mouse platelet ATP secretion was measured as a surrogate marker for dense granule secretion. Mouse platelets treated with DPAn-6 or oxylipins prior to incubation with CHRONO-LUME for 2 minutes. Platelets were stimulated with the indicated agonists, and luminescence was measured over time in a Lumi-Aggregometer (CHRONO-LOG Model 700D).
Flow cytometry
Mouse platelets (1.5 × 107) were treated with DPAn-6 and/or its oxylipins were stained with fluorescein isothiocyanate–conjugated CD62P antibody (emfret ANALYTICS, Eibelstadt, Germany) and stimulated with thrombin for P-selectin expression. Platelets were fixed with 2% paraformaldehyde for 10 minutes and diluted in Tyrode’s buffer. P-selectin surface expression was analyzed using a ZE5 Cell Analyzer (Bio-Rad).
Vasodilator-stimulated phosphoprotein phosphorylation
Human and mouse platelets (3 × 108) were treated with PUFAs or oxylipins for 1 minute and quenched in 5× sample buffer. Samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted for p-VASP Antibody (Ser 239) (Santa Cruz Biotechnology), anti-VASP (phospho S157) antibody; and total VASP (Enzo Life Sciences).
Cyclic adenosine monophosphate assay
Human platelets were treated with 3-isobutyl-1-methylxanthine (IBMX) for 30 minutes at room temperature, followed by PUFAs or oxylipins for 1 minute. Reactions were quenched, and cyclic adenosine monophosphate (cAMP) enzyme-linked immunosorbent assay (Enzo Life Sciences) was performed.
PPAR transactivation assay
PPARα activation was measured as previously described.18,19 HEK293T cells were cotransfected with 60 ng of CMX-Gal4-mPPARα, 180 ng of UAS reporter Tk-MH100x4-luc, and 250 ng of CMX–β-galactosidase and maintained for 24 hours. Compounds were added in a log–dose-dependent manner, incubated for 8 hours, lysed, and analyzed for luciferase and β-galactosidase activities. PPAR and luciferase constructs were a gift from Steven A. Kliewer (UT Southwestern Medical Center, Dallas, TX).
Ex vivo microfluidic perfusion chamber assays
Microfluidic perfusion chambers (μ-slide VI0.1; Ibidi) were coated with 100 μg/mL collagen (CHRONO-LOG) in 1× phosphate-buffered saline overnight at 4°C. Whole blood collected in sodium citrate vacutainer tubes was treated with PUFA or oxylipins and 1 μM 3,3′-dihexyloxacarbocyanine iodide (Thermo Fisher Scientific) for 10 minutes and immediately recalcified with 1 mM CaCl2 prior to perfusion at arterial shear of 1800/s for 5 minutes. Platelet accumulation was monitored using an inverted fluorescent microscope (Zeiss Axio Observer Z1 Marianas; 20× objective) and analyzed with SlideBook 6.0 software (3i Intelligent Imaging Innovations).
Laser-induced cremaster arteriole thrombosis model
Male mice (8-10 weeks of age) were anesthetized by intraperitoneal injection of ketamine/xylazine (100 mg/kg), and a tracheal tube was inserted to facilitate breathing.10,17 A jugular vein catheter was established to inject antibodies and reagents. Anti-platelet (DyLight 488 anti-GPIb; 1 μg/g) and anti-fibrin (Alexa Fluor 647; 0.3 μg/g) antibodies were administered prior to intravital microscopy. Compounds were administered for 10 minutes prior to induction of vascular injury using a laser ablation system (Ablate! photoablation system; 3i). Images were acquired in real-time, using a 63× water-immersion objective, with a Zeiss Axio Examiner Z1 multichannel fluorescent microscope equipped with a solid laser launch system and high-speed sCMOS camera. Images were analyzed using SlideBook software.
Tail bleeding assay
Mice were anesthetized with ketamine/xylazine, and the tip of the tail (5 mm) was excised with a sterile scalpel. The tail was immersed in saline solution (0.9%), and bleeding time was assessed.
Statistics
Paired 2-tailed Student t tests, 1- and 2-way analysis of variance (ANOVA), and 2 factor mixed-effects model were performed with Prism 8 (GraphPad Software) to analyze the data. Multiple statistical analyses were used in this study; the statistical test used in each assay is reported in the figure legends. Data are means ± standard error of the mean (SEM).
Results
12-LOX kinetics with fatty acid substrates
The in vitro production of DPA-derived oxylipins by recombinant h12-LOX was examined to enable biosynthetic flux determination. The major in vitro products were similar to the ex vivo platelet-derived results (Table 1), with the omega-9 product (14-HpDPA), and not the omega-12 product (11-HpDPA), being the major product. This dual positional specificity (ie, omega-9 and omega-12 products) is unusual because only the omega-9 product (ie, the 12-oxylipin) is observed when arachidonic acid (AA) and eicosapentaenoic acid (EPA) are the substrates. These results are most likely due to a difference in active site binding of the 22-carbon fatty acids docosahexaenoic acid (DHA) and DPA compared with the 20-carbon fatty acids AA and EPA.20 h12-LOX produced ∼20% of the 11C product (omega-12 oxygenation) with DHA and DPAn-3 as substrates, but DPAn-6 manifested significantly less 11C product (∼2%) (Table 1). These results suggest that DHA and DPAn-3, which have an omega-3 double bond, may insert deeper into the 12-LOX active site, resulting in a greater percentage of the 11C product.
The catalytic efficiency of recombinant 12-LOX with DHA, DPAn-3, and DPAn-6 was investigated to understand the biological relevancy in comparison with the canonical fatty acid substrate AA (Table 2). The fatty acid turnover rate (kcat) values for DHA, DPAn-3, and DPAn-6 are all within twofold of that for AA, indicating comparable rates for product release.21 The catalytic efficiency of the enzymes (kcat/KM) for DHA, DPAn-3, and DPAn-6 are within threefold of that for AA, indicating similar rates for substrate capture, as well. These data indicate that the increased length and unsaturation for these fatty acids (DHA, DPAn-3, and DPAn-6) has a limited effect on enzyme catalysis compared with AA.
Dietary supplementation with omega-6 PUFA affects platelet lipid content
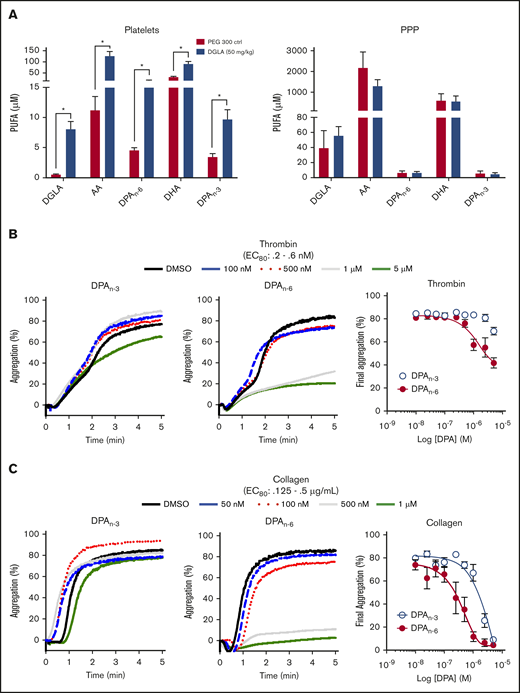
Previously, acute IV administration of DGLA was shown to result in reduced thrombi formation in vivo through a 12-LOX–dependent mechanism.9,10 Although much of the observed antiplatelet effect was shown to be due to DGLA and its oxylipin, the possibility of other omega-6 PUFAs due to endogenous desaturates following dietary supplementation, had not been considered.22 To investigate the importance of lipid products of DGLA, mice were supplemented orally daily with 50 mg/kg DGLA or vehicle control for 1 month. Following completion of the oral gavage treatment, washed platelets were collected and assessed for a subset of omega-3 and omega-6 PUFAs (DGLA, AA, DPA, and DHA). Omega-3 and omega-6 forms of DPA (DPAn-3 and DPAn-6, respectively), AA, and DGLA were significantly increased in platelets, but not in platelet-poor plasma (Figure 1A), confirming previous reports that habitual feeding of PUFAs leads to increased PUFA incorporation into membranes.23,24 Surprisingly, the largest increase in a fatty acid in the membrane was from DPAn-3 and DPAn-6. To determine whether DPAn-3 and DPAn-6 fatty acids play a regulatory role in platelet activation, each fatty acid was dose dependently added to washed human platelets (10 nM-5 μM) at a concentration well below the observed concentration of DPA in humans following supplementation prior to the 80% effective concentration (EC80) agonist stimulation with thrombin (Figure 1B) or collagen (Figure 1C).25 Fatty acid effects on platelet function were assessed by platelet aggregation. Human platelets treated with DPAn-6 showed markedly reduced platelet aggregation in response to thrombin or collagen compared with DPAn-3. For this reason, DPAn-6 and its corresponding oxylipins were evaluated further.
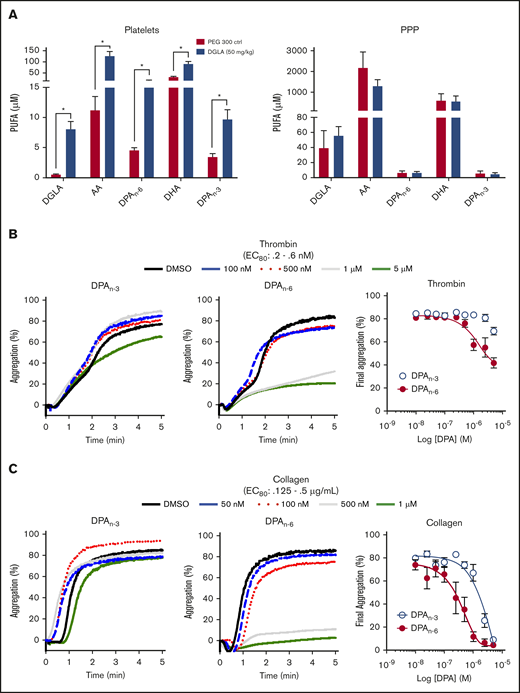
Dietary supplementation of DGLA enhances DPAn-3and DPAn-6in mouse platelets. (A) Platelets (left panel) and platelet-rich plasma (PPP; right panel) from WT mice were measured for omega-3 and omega3 PUFAs (DGLA, AA, DPAn-6, DPAn-3, and DHA) following 1 month of daily dietary supplementation of 50 mg/kg of DGLA (n = 5) or polyethylene glycol 300 control (n = 3). The potency of DPAn-3 and DPAn-6 was determined by human platelets measuring aggregation in response to thrombin (B) or collagen (C) (n = 10). Data are means ± SEM. *P < .05, 1-way ANOVA.
Dietary supplementation of DGLA enhances DPAn-3and DPAn-6in mouse platelets. (A) Platelets (left panel) and platelet-rich plasma (PPP; right panel) from WT mice were measured for omega-3 and omega3 PUFAs (DGLA, AA, DPAn-6, DPAn-3, and DHA) following 1 month of daily dietary supplementation of 50 mg/kg of DGLA (n = 5) or polyethylene glycol 300 control (n = 3). The potency of DPAn-3 and DPAn-6 was determined by human platelets measuring aggregation in response to thrombin (B) or collagen (C) (n = 10). Data are means ± SEM. *P < .05, 1-way ANOVA.
The antiplatelet effects of 12-LOX products of DPAn-6
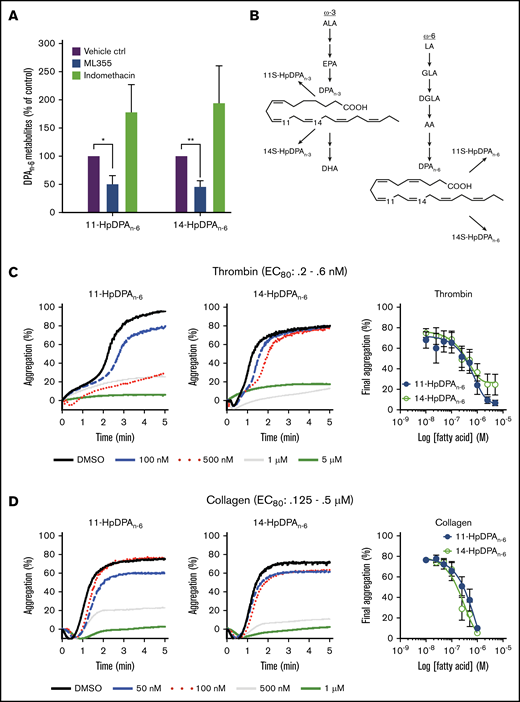
As previously described, the antithrombotic effects of DGLA are partially due to its 12-LOX oxylipin.8,10,26 To determine which oxygenase is responsible for the generation of the DPA-derived oxylipins, platelets were treated with DPAn-6 and predominantly produced 14-HpDPAn-6, with a small amount of 11-HpDPAn-6 (Table 1). This ratio is consistent with the in vitro data (Figure 2), as well as with 12-LOX being the source of these oxylipins. Subsequently, platelets exposed to DPAn-6 were exposed to the COX-1 inhibitor, indomethacin, or the 12-LOX inhibitor, ML-355. 14-HpDPAn-6 was the predominant oxylipin compared with 11-HpDPAn-6 in platelets treated with DPAn-6 (Figure 2A; Table 1); however, the levels of both oxylipins were reduced with ML-355 (Figure 2A). Surprisingly, although thromboxane and prostaglandin E2 were observed, suggesting that COX-1–derived AA products were present (data not shown), no COX-1–derived products of DPAn-6 were observed in platelet samples treated with excess DPAn-6 at the threshold of detection with LC-MS/MS (10 pg/9 × 108 platelets), suggesting that DPAn-6 is an inert substrate of COX-1, whereas, 12-LOX–derived oxylipins of DPAn-6 appear to be the lipid mediators exerting a regulatory role on platelet function (Figure 2B).
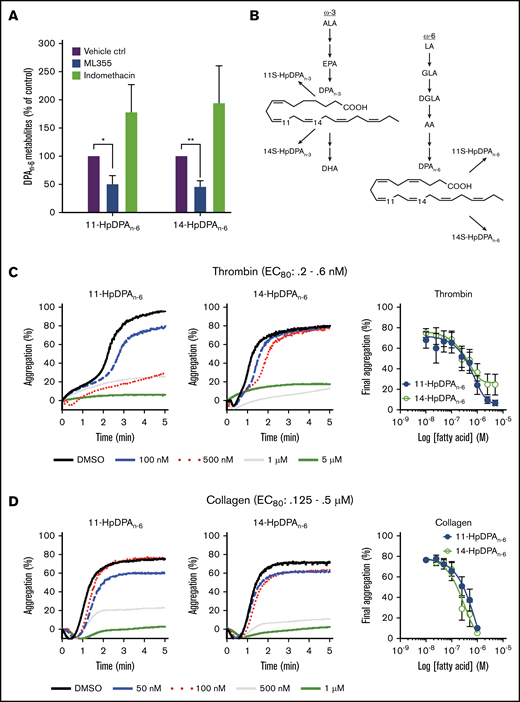
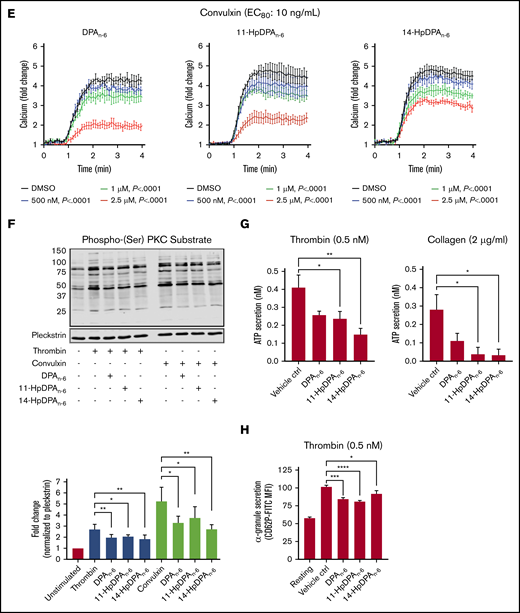
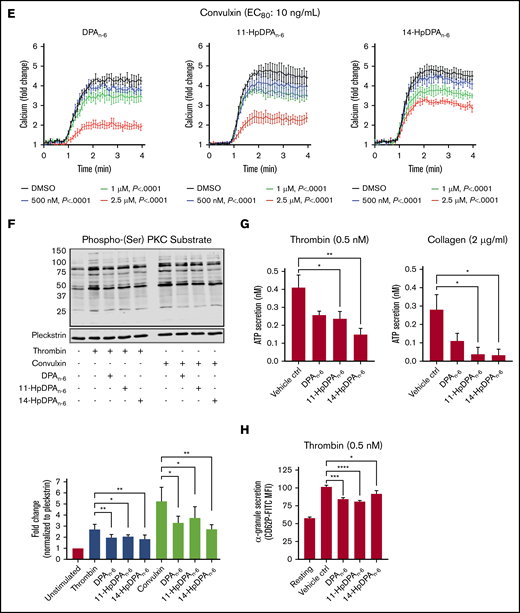
12-LOX-derived DPAn-6and oxylipins 11-HpDPAn-6and 14-HpDPAn-6inhibit platelet activation. (A) Oxylipins derived from platelets (n = 4) were assessed by incubating human platelets with 10 μM DPA in the presence of vehicle control (ctrl), 12-LOX inhibitor (ML355), or COX-1 inhibitor (indomethacin) prior to mass spectrometry. (B) The major metabolites produced from DPAn-6 in platelets are 12-LOX–derived oxylipins, 11-HpDPAn-6, and 14-HpDPAn-6. The effects of 11-HpDPAn-6 and 14-HpDPAn-6 on platelet activity were assessed by incubating human platelets with increasing concentrations of DPAn-6 metabolites for 5 minutes prior to stimulation with thrombin (n = 10) (C) or collagen (n = 10) (D). (E) Calcium mobilization induced by the glycoprotein VI agonist convulxin was assessed in the absence or presence of DPAn-6 (left panel), 11-HpDPAn-6 (middle panel), or 14-HpDPAn-6 (right panel) (n = 5). (F) PKC activity was assessed in the absence or presence of DPAn-6 (2.5 μΜ), 11-HpDPAn-6, (2.5 μΜ), or 14-HpDPAn-6 (2.5 μΜ) prior to stimulation with 0.2 nM thrombin (n = 4) or 10 ng/mL convulxin (n = 4). (G) Platelet ATP secretion was measured in the absence or presence of DPAn-6 (1 μM), 11-HpDPAn-6 (1 μM), or 14-HpDPAn-6 (1 μM) prior to stimulation with 0.5 nM thrombin (left panel; n = 6) or 2 μg/mL collagen (right panel; n = 4). (H) α-Granule secretion was assessed in the absence or presence of DPAn-6 (1 μM), 11-HpDPAn-6 (1 μM), or 14-HpDPAn-6 (1 μM) prior to stimulation with thrombin 0.5 nM (n = 5). Data are means ± standard deviation (SD) for PKC activation means ± SEM for others. *P < .05, **P < .01, ***P < .001, ****P < .0001, 1- or 2-way ANOVA. DMSO, dimethyl sulfoxide.
12-LOX-derived DPAn-6and oxylipins 11-HpDPAn-6and 14-HpDPAn-6inhibit platelet activation. (A) Oxylipins derived from platelets (n = 4) were assessed by incubating human platelets with 10 μM DPA in the presence of vehicle control (ctrl), 12-LOX inhibitor (ML355), or COX-1 inhibitor (indomethacin) prior to mass spectrometry. (B) The major metabolites produced from DPAn-6 in platelets are 12-LOX–derived oxylipins, 11-HpDPAn-6, and 14-HpDPAn-6. The effects of 11-HpDPAn-6 and 14-HpDPAn-6 on platelet activity were assessed by incubating human platelets with increasing concentrations of DPAn-6 metabolites for 5 minutes prior to stimulation with thrombin (n = 10) (C) or collagen (n = 10) (D). (E) Calcium mobilization induced by the glycoprotein VI agonist convulxin was assessed in the absence or presence of DPAn-6 (left panel), 11-HpDPAn-6 (middle panel), or 14-HpDPAn-6 (right panel) (n = 5). (F) PKC activity was assessed in the absence or presence of DPAn-6 (2.5 μΜ), 11-HpDPAn-6, (2.5 μΜ), or 14-HpDPAn-6 (2.5 μΜ) prior to stimulation with 0.2 nM thrombin (n = 4) or 10 ng/mL convulxin (n = 4). (G) Platelet ATP secretion was measured in the absence or presence of DPAn-6 (1 μM), 11-HpDPAn-6 (1 μM), or 14-HpDPAn-6 (1 μM) prior to stimulation with 0.5 nM thrombin (left panel; n = 6) or 2 μg/mL collagen (right panel; n = 4). (H) α-Granule secretion was assessed in the absence or presence of DPAn-6 (1 μM), 11-HpDPAn-6 (1 μM), or 14-HpDPAn-6 (1 μM) prior to stimulation with thrombin 0.5 nM (n = 5). Data are means ± standard deviation (SD) for PKC activation means ± SEM for others. *P < .05, **P < .01, ***P < .001, ****P < .0001, 1- or 2-way ANOVA. DMSO, dimethyl sulfoxide.
11-HpDPAn-6 and 14-HpDPAn-6 products from DPAn-6 were assessed for their ability to regulate agonist-induced platelet aggregation (Figure 2C-D). 11-HpDPAn-6 and 14-HpDPAn-6 dose dependently (50 nM-5 μM) inhibited platelet aggregation in response to the EC80 concentration of thrombin (Figure 2C) or collagen (Figure 2D). Additionally, intracellular calcium mobilization, a precursor to integrin-mediated platelet aggregation, was measured (Figure 2E). All agonists dose dependently decreased calcium mobilization induced by EC80 concentration of convulxin, a snake venom that directly activates the glycoprotein VI collagen receptor (Figure 2E). In line with the observed attenuation of calcium mobilization, PKC activity and dense granule secretion induced by EC80 concentration of thrombin or convulxin were attenuated (Figure 2F-G). Similarly, thrombin-mediated α-granule secretion was reduced in mouse platelets treated with DPAn-6 or its oxylipins compared with the vehicle control (Figure 2H). The maximal inhibitory concentration required for both oxylipins to inhibit platelet aggregation was <1 μM for collagen and <5 μM for thrombin-stimulated platelet aggregation.
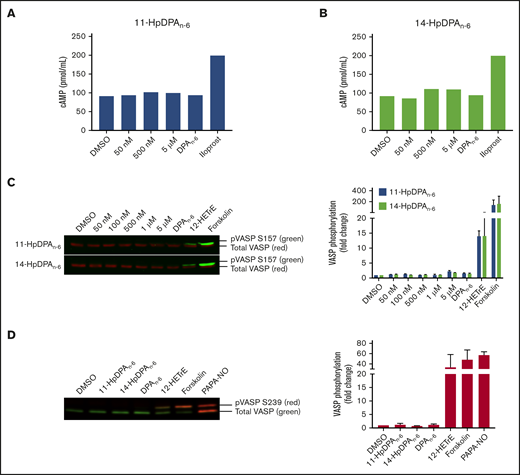
12-LOX–derived oxylipins of DPAn-6, 11-HpDPAn-6, and 14-HpDPAn-6 inhibit platelet function in a cAMP-independent and cyclic guanosine 3′,5′-cyclic monophosphate–independent manner
The 12-LOX–derived oxylipin of DGLA, 12(S)-HETrE, was previously shown to inhibit platelet activation and thrombosis through a Gαs-dependent mechanism, coupled to the prostacyclin receptor.9,10 To determine whether the oxylipins of DPAn-6 inhibit platelet activation in a similar mechanism to 12(S)-HETrE, intracellular cAMP was measured in washed human platelets incubated with a nonselective phosphodiesterase inhibitor, IBMX (10 μM), prior to treatment with increasing concentrations of 11-HpDPAn-6, 14-HpDPAn-6, or DPAn-6 (Figure 3A). As expected, platelets treated with iloprost, a synthetic analog of prostacyclin, enhanced cAMP generation; however, cAMP production was not induced at serine 157 (S157) in platelets treated with 11-HpDPAn-6, 14-HpDPAn-6, or DPAn-6 (Figure 3A-B). Additionally, phosphorylation of vasodilator-stimulated phosphoprotein (VASP) at S157, a surrogate marker for active cAMP-activated protein kinase (protein kinase A), was measured in the presence of DPAn-6 or its 12-LOX metabolites for 1 minute. Platelets treated with 12(S)-HETrE or forskolin, a direct activator of adenylyl cyclase, showed enhanced S157 VASP phosphorylation; however, platelets treated with DPAn-6 or its 12-LOX oxylipins did not show VASP phosphorylation (Figure 3C). Phosphorylation of VASP at serine 239 (S239), the selective protein kinase G phosphorylation site, was also assessed in the presence of DPAn-6 or its 12-LOX oxylipins for 1 minute. Neither DPAn-6 nor its 12-LOX oxylipins induced VASP phosphorylation at S239 compared with the positive control, PAPA NONOate, an NO donor (Figure 3D).
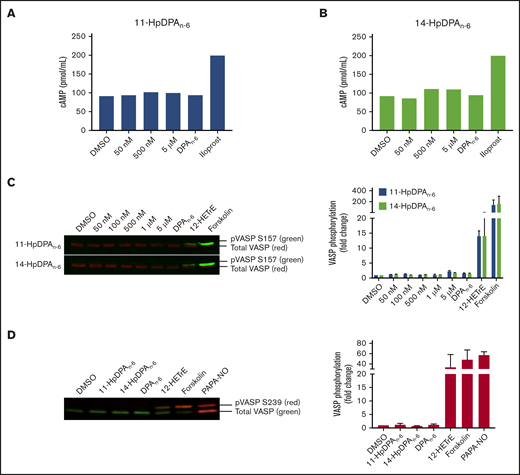
12-LOX–derived DPAn-6oxylipins, 11-HpDPAn-6and 14-HpDPAn-6, do not impinge on the Gαssignaling pathway in platelets. Washed human platelets were treated with increasing concentrations of 11-HpDPAn-6 (A) or 14-HpDPAn-6 (B), along with DPAn-6 (10 μM) or iloprost (1 μM) for 1 minute in the presence of IBMX (10 μM) (n = 2). (C) Human platelets (n = 5) were treated with dimethyl sulfoxide (DMSO), increasing concentrations of 11-HpDPAn-6 or 14-HpDPAn-6, DPAn-6 (10 μM), 12(S)-HETrE (25 μM), or forskolin (1 μM) for 1 minute and immediately lysed, resolved on SDS-PAGE gel, and immunoblotted for VASP phosphorylation at S157 or total VASP proteins. Data are means ± SEM. (D) Mouse platelets (n = 3) were treated with DMSO, 11-HpDPAn-6 (1 μM), 14-HpDPAn-6 (1 μM), DPAn-6 (10 μM), 12(S)-HETrE (25 μM), forskolin (1 μM), or PAPA NONOate (5 μM) for 1 minute and immediately lysed, resolved on SDS-PAGE gel, and immunoblotted for VASP phosphorylation at S239 or total VASP proteins. Data are means ± SD. VASP phosphorylation levels were normalized to total VASP and DMSO control level to estimate the fold change.
12-LOX–derived DPAn-6oxylipins, 11-HpDPAn-6and 14-HpDPAn-6, do not impinge on the Gαssignaling pathway in platelets. Washed human platelets were treated with increasing concentrations of 11-HpDPAn-6 (A) or 14-HpDPAn-6 (B), along with DPAn-6 (10 μM) or iloprost (1 μM) for 1 minute in the presence of IBMX (10 μM) (n = 2). (C) Human platelets (n = 5) were treated with dimethyl sulfoxide (DMSO), increasing concentrations of 11-HpDPAn-6 or 14-HpDPAn-6, DPAn-6 (10 μM), 12(S)-HETrE (25 μM), or forskolin (1 μM) for 1 minute and immediately lysed, resolved on SDS-PAGE gel, and immunoblotted for VASP phosphorylation at S157 or total VASP proteins. Data are means ± SEM. (D) Mouse platelets (n = 3) were treated with DMSO, 11-HpDPAn-6 (1 μM), 14-HpDPAn-6 (1 μM), DPAn-6 (10 μM), 12(S)-HETrE (25 μM), forskolin (1 μM), or PAPA NONOate (5 μM) for 1 minute and immediately lysed, resolved on SDS-PAGE gel, and immunoblotted for VASP phosphorylation at S239 or total VASP proteins. Data are means ± SD. VASP phosphorylation levels were normalized to total VASP and DMSO control level to estimate the fold change.
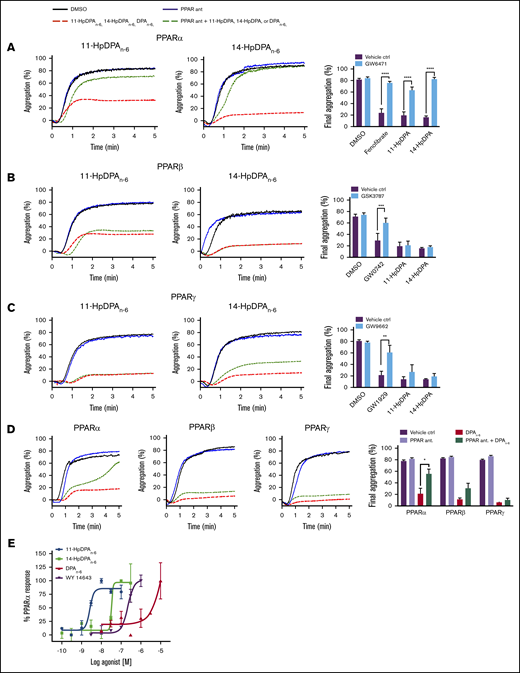
12-LOX–derived oxylipins of DPAn-6 modulate platelet activation through PPAR
Oxylipins have been shown to activate an intracellular receptor known as PPAR, a subfamily of ligand-inducible transcription factors.26-29 Because there is no nucleus in the platelet, PPAR activation would not be expected to have any effect on platelet reactivity; however, the reported nuclear-independent signaling effects of some of the members of the PPAR family in the platelet suggest the possibility that DPAn-6 oxylipins regulate platelet reactivity in this manner.30,31 Therefore, DPAn-6 and its 12-LOX–derived oxylipins were assessed for their ability to directly activate PPARs in a nongenomic manner in platelets.
Mammals express 3 isoforms of PPAR: α, β/δ, and γ. To determine which PPAR isoform is activated by DPAn-6 or its oxylipins, selective antagonists for each PPAR isoform were tested to determine PPAR isoform selectivity (Figure 4). Prior to incubation with 11-HpDPAn-6 or 14-HpDPAn-6, or known PPAR agonists, washed human platelets were treated or not with the following PPAR antagonist: GW6471 for PPARα (Figure 4A), GSK3787 for PPARβ/δ (Figure 4B), or GW9662 for PPARγ (Figure 4C). Selective PPAR agonists, fenofibrate (PPARα), GW0742 (PPARβ/δ), and GW1929 (PPARγ) significantly inhibited collagen-stimulated platelet aggregation (Figure 4A-C), as previously reported32-34 ; however, when pretreated with their respective PPAR antagonist, the PPAR agonists were unable to inhibit platelet aggregation. Although pretreatment with GW6471 (PPARα inhibitor) rescued oxylipin-mediated inhibition of platelet aggregation, platelets treated with GSK3787 or GW9662 had no effect on DPAn-6, 11-HpDPAn-6, or 14-HpDPAn-6 inhibition of platelet aggregation.
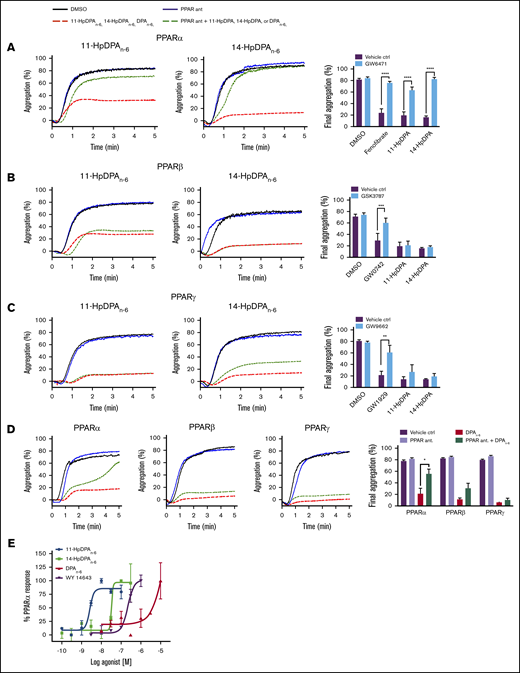
12-LOX–derived DPAn-6oxylipins, 11-HpDPAn-6and 14-HpDPAn-6, modulate platelet activation through PPAR. Washed human platelets were treated with 10 μM GW6471 (PPARα antagonist) (n = 4) (A), 10 μM GSK3787 (PPARβ antagonist) (n = 4) (B), or 5 μM GW9962 (PPARγ antagonist) (n = 4) (C) for 5 minutes prior to 0.5 to 1 μM incubation with 11-HpDPAn-6 or 14-HpDPAn-6 and platelet aggregation stimulation with EC80 collagen. (D) Similarly, platelets were treated with 1 to 2.5 μM DPAn-6 in the presence of PPAR antagonists prior to collagen-induced aggregation (n = 4). Representative aggregation tracings for 11-HpDPAn-6, 14-HpDPAn-6, and DPAn-6 are shown on the left. (E) A cell-based PPAR transactivation reporter assay was performed in HEK293T cells that had been dose dependently treated with DPAn-6, 11-HpDPAn-6, 14-HpDPAn-6, or PPARα agonist WY 14643 for 8 hours prior to PPARα luciferase assay. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < .001, ****P < .0001, 1-way statistical tests.
12-LOX–derived DPAn-6oxylipins, 11-HpDPAn-6and 14-HpDPAn-6, modulate platelet activation through PPAR. Washed human platelets were treated with 10 μM GW6471 (PPARα antagonist) (n = 4) (A), 10 μM GSK3787 (PPARβ antagonist) (n = 4) (B), or 5 μM GW9962 (PPARγ antagonist) (n = 4) (C) for 5 minutes prior to 0.5 to 1 μM incubation with 11-HpDPAn-6 or 14-HpDPAn-6 and platelet aggregation stimulation with EC80 collagen. (D) Similarly, platelets were treated with 1 to 2.5 μM DPAn-6 in the presence of PPAR antagonists prior to collagen-induced aggregation (n = 4). Representative aggregation tracings for 11-HpDPAn-6, 14-HpDPAn-6, and DPAn-6 are shown on the left. (E) A cell-based PPAR transactivation reporter assay was performed in HEK293T cells that had been dose dependently treated with DPAn-6, 11-HpDPAn-6, 14-HpDPAn-6, or PPARα agonist WY 14643 for 8 hours prior to PPARα luciferase assay. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < .001, ****P < .0001, 1-way statistical tests.
To validate the direct interaction of PPARα with DPAn-6 oxylipins (Figure 4A-D), a cell-based PPAR transactivation reporter assay was conducted to determine the potency of PPARα binding to DPAn-6 and its 12-LOX oxylipins in HEK293T cells transiently cotransfected with CMX-Gal4-PPARα, CMX-4–Gal4–β-galactosidase, and Tk-MH100x40luc.18,19 11-HpDPAn-6 (EC50 = 2.62 × 10−9 M) and 14-HpDPAn-6 (EC50 = 5.58 × 10−8 M) dose dependently and potently bind to PPARα and induce activation (Figure 4E). As expected, WY 14643, a known PPARα agonist, activated PPARα in HEK293T cells (EC50 = 2.25 × 10−7 M). Altogether, these data strongly implicate 11-HpDPAn-6 and 14-HpDPAn-6 as negative regulators of platelet function through activation of PPARα in platelets at nanomolar concentrations.
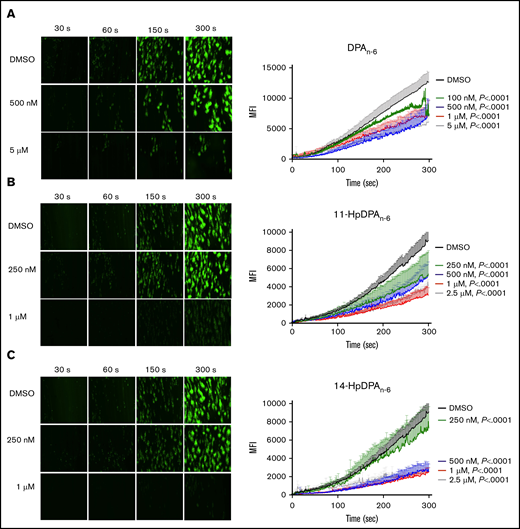
11-HpDPAn-6 and 14-HpDPAn-6 attenuate platelet adhesion under arterial shear conditions
To determine whether the inhibitory effects of DPAn-6 and its 12-LOX oxylipins could be recapitulated in a more physiological setting, platelet adhesion was assessed ex vivo in human whole blood under arterial flow conditions in a microfluidic chamber. Whole blood collected in sodium citrate tubes was recalcified and treated with varying concentrations of DPAn-6 or 12-LOX oxylipins for 5 minutes prior to perfusion over a collagen-coated surface at an arterial shear rate (1800/s). Vehicle-treated platelets labeled with calcein-AM accumulated rapidly and adhered on the collagen-coated surface (Figure 5). Increasing concentrations of DPAn-6 or its oxylipins significantly reduced platelet adherence.
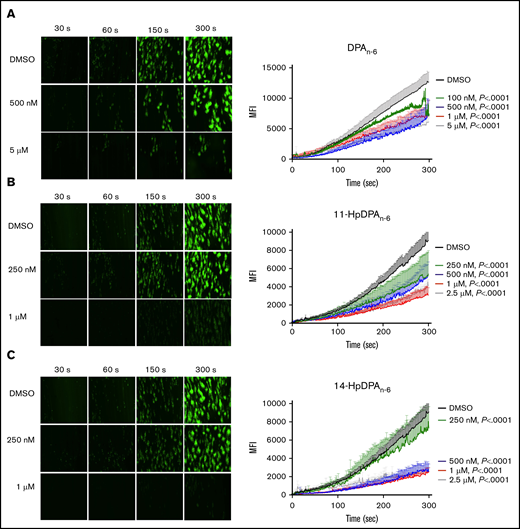
12-LOX–derived DPAn-6oxylipins, 11-HpDPAn-6and 14-HpDPAn-6, inhibit human platelet accumulation on a collagen-coated surface under arterial shear flow conditions. Whole blood collected in a sodium-citrate vacutainer tube was incubated with dimethyl sulfoxide (DMSO) or 100 nM to 5 μM DPAn-6 (n = 6) or 250 nM to 2.5 μM DPAn-6 (n = 5) (A), 11-HpDPAn-6 (n = 5) (B), or 14-HpDPAn-6 (n = 5) (C) for 5 minutes at 37°C prior to perfusion over a collagen-coated surface at arterial shear rate (1800/s) for 5 minutes (original magnification, ×20). Representative figures for each condition are shown to the left of each composite graph. Data are mean ± SEM. Statistical significance calculated using 2-way ANOVA. MFI, mean fluorescence intensity.
12-LOX–derived DPAn-6oxylipins, 11-HpDPAn-6and 14-HpDPAn-6, inhibit human platelet accumulation on a collagen-coated surface under arterial shear flow conditions. Whole blood collected in a sodium-citrate vacutainer tube was incubated with dimethyl sulfoxide (DMSO) or 100 nM to 5 μM DPAn-6 (n = 6) or 250 nM to 2.5 μM DPAn-6 (n = 5) (A), 11-HpDPAn-6 (n = 5) (B), or 14-HpDPAn-6 (n = 5) (C) for 5 minutes at 37°C prior to perfusion over a collagen-coated surface at arterial shear rate (1800/s) for 5 minutes (original magnification, ×20). Representative figures for each condition are shown to the left of each composite graph. Data are mean ± SEM. Statistical significance calculated using 2-way ANOVA. MFI, mean fluorescence intensity.
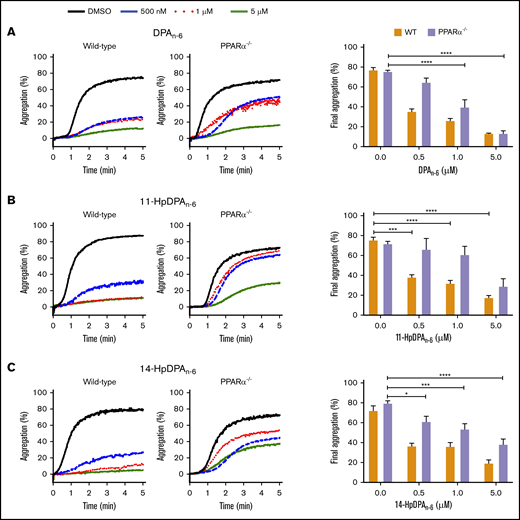
11-HpDPAn-6 and 14-HpDPAn-6 are unable to sufficiently inhibit platelet aggregation in the absence of PPARα
To further assess the roles of 11-HpDPAn-6 and 14-HpDPAn-6 as potential ligands for PPARα, platelets from PPARα−/− mice, which were previously reported to be nonresponsive to fenofibrate following collagen stimulation,32 were used for subsequent ex vivo and in vivo assays. Platelets from WT or PPARα−/− mice were treated with 0.5, 1, or 5 μM DPAn-6, 11-HpDPAn-6, or 14-HpDPAn-6 for 5 minutes prior to EC80 collagen-induced aggregation (Figure 6). A significant difference in aggregation in response to 0.5 μM DPAn-6 was observed in platelets from WT and PPARα−/− mice; collagen-mediated platelet aggregation was not inhibited in the latter (Figure 6A). Additionally, 0.5 and 1 μM of 11-HpDPAn-6 and 14-HpDPAn-6 failed to fully inhibit platelets from PPARα−/− mice in response to collagen compared with WT mice treated with the same concentrations (Figure 6B-C). Altogether, the pharmacological and genetic inhibition and ablation, as well as cell-based assays, demonstrate the selectivity of 11-HpDPAn-6 and 14-HpDPAn-6 for PPARα activation over other isoforms of PPAR at nanomolar range fatty acid and metabolite levels.
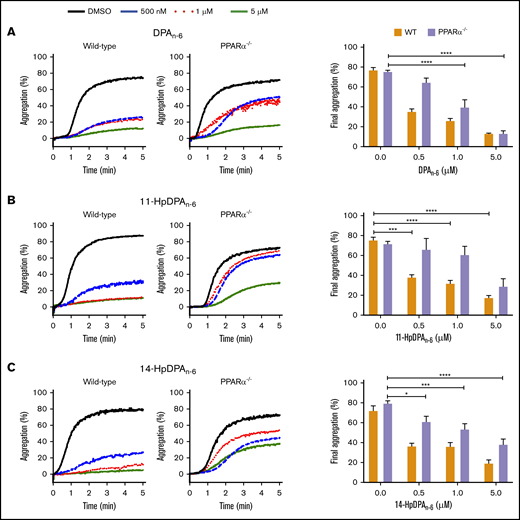
PPARα mediates the inhibitory effects of DPAn-6metabolites on platelet aggregation. Washed platelets from WT and PPARα−/− mice were incubated with DPAn-6 (A), 11-HpDPAn-6 (B), or 14-HpDPAn-6 (C), ranging from 0.5 to 5 μM, for 5 minutes prior to EC80 collagen-induced aggregation. Data are means ± SEM. *P < .05, ***P < .001, ****P < .0001, 1-way ANOVA.
PPARα mediates the inhibitory effects of DPAn-6metabolites on platelet aggregation. Washed platelets from WT and PPARα−/− mice were incubated with DPAn-6 (A), 11-HpDPAn-6 (B), or 14-HpDPAn-6 (C), ranging from 0.5 to 5 μM, for 5 minutes prior to EC80 collagen-induced aggregation. Data are means ± SEM. *P < .05, ***P < .001, ****P < .0001, 1-way ANOVA.
11-HpDPAn-6 and 14-HpDPAn-6 inhibit thrombus growth in vivo in a PPARα-dependent manner
To determine whether the antiplatelet effects of DPAn-6 and its 12-LOX oxylipins contribute to the inhibition of thrombus formation and growth in vivo, a laser-induced cremaster arteriole thrombosis model was applied to male mice, as previously described.10 The effects of DPAn-6 and its 12-LOX oxylipins on platelet accumulation and fibrin formation following vascular injury were determined in WT mice pretreated with 1 mg/kg DPAn-6, 11-HpDPAn-6, 14-HpDPAn-6, or vehicle (Figure 7). As demonstrated in representative images of thrombus growth in Figure 7A (supplemental Video 1), laser-induced thrombus formation in the cremaster arterioles of mice was significantly inhibited compared with WT mice following IV administration of DPAn-6, 11-HpDPAn-6, or 14-HpDPAn-6. Interestingly, administration of 11-HpDPAn-6 or 14-HpDPAn-6 inhibited the extent of thrombosis in mice more potently than did DPA treatment. This observation was further supported by analysis of dynamic platelet recruitment in growing thrombi (Figure 7B). Although DPAn-6, 11-HpDPAn-6, and 14-HpDPAn-6 all inhibit platelet recruitment to the thrombus, their effects on fibrin formation differed (Figure 7C). Fibrin formation was strongly inhibited following administration of DPAn-6. In contrast, fibrin formation was slightly delayed in mice treated with 11-HpDPAn-6, and 14-HpDPAn-6 treatment potently inhibited fibrin formation compared with the other conditions tested.
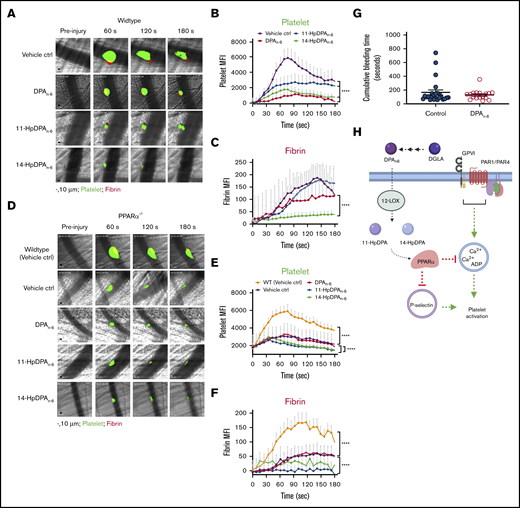
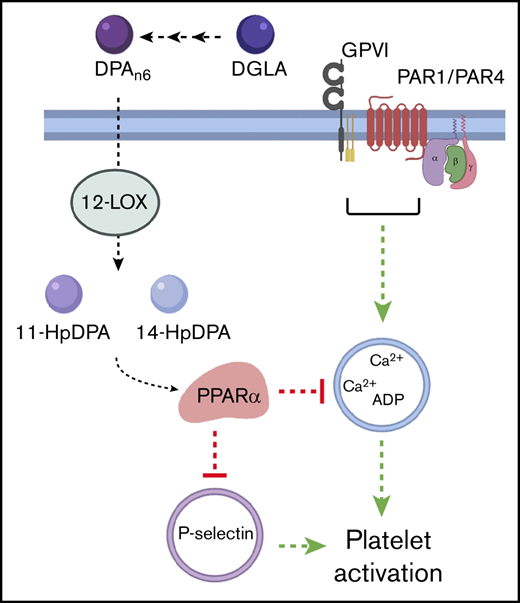
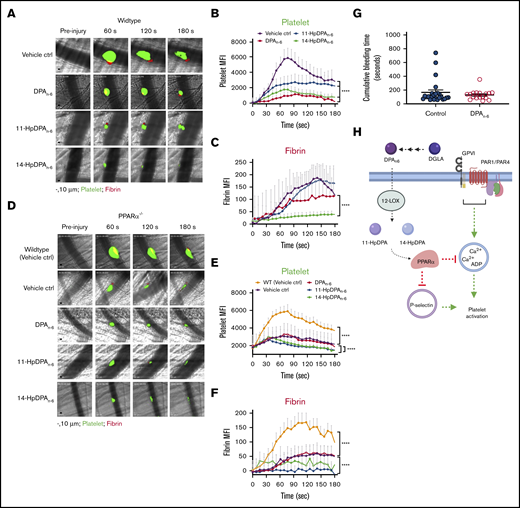
DPAn-6and its oxylipins, 11-HpDPAn-6and 14-HpDPAn-6, impair in vivo thrombus formation in laser-induced cremaster arteriole thrombosis models. (A) Representative images of platelet accumulation (green) and fibrin formation (red) in growing thrombi in cremaster arterioles in WT control treated with vehicle control (ctrl), DPAn-6, 11-HpDPAn-6, or 14-HpDPAn-6. Time after vascular injury is indicated above (n = 3 mice, 10-12 thrombi per mouse). Scale bars, 10 μm. (B) Dynamics of platelet accumulation in thrombi analyzed by change in fluorescent intensity following injury. (C) Dynamics of fibrin formation within thrombi analyzed by change in fluorescent intensity following injury. (D) Representative images of platelet accumulation (green) and fibrin formation (red) in growing thrombi in cremaster arterioles in PPARα−/− mice treated with vehicle control (ctrl), DPAn-6, 11-HpDPAn-6, or 14-HpDPAn-6. Time after vascular injury is indicated above (n = 3 mice, 10-12 thrombi per mouse). (E) Dynamics of platelet accumulation in thrombi in PPARα−/− mice analyzed by change in fluorescent intensity. (F) Dynamics of fibrin formation in thrombi in PPARα−/− mice analyzed by change in fluorescent intensity following injury. (G) Mean tail bleeding of control mice (n = 24) or mice treated with DPAn-6 (n = 17) is denoted by the horizontal line. (H) The proposed model of DPAn-6 12-LOX oxylipins’ inhibitory effect on platelet activation. DPAn-6 12-LOX oxylipins derived from the elongation of DGLA into the bioactive lipids 11-HpDPAn-6 and 14-HpDPAn-6. DPAn-6 oxylipins regulate platelet function through activation of PPARα, which leads to inhibition of dense and α-granule release, calcium mobilization, and platelet reactivity in response to G protein–coupled receptor–based or immunoreceptor tyrosine-based activation motif–mediated platelet activation. Data are means ± SEM. ****P < .0001, 2-way ANOVA.
DPAn-6and its oxylipins, 11-HpDPAn-6and 14-HpDPAn-6, impair in vivo thrombus formation in laser-induced cremaster arteriole thrombosis models. (A) Representative images of platelet accumulation (green) and fibrin formation (red) in growing thrombi in cremaster arterioles in WT control treated with vehicle control (ctrl), DPAn-6, 11-HpDPAn-6, or 14-HpDPAn-6. Time after vascular injury is indicated above (n = 3 mice, 10-12 thrombi per mouse). Scale bars, 10 μm. (B) Dynamics of platelet accumulation in thrombi analyzed by change in fluorescent intensity following injury. (C) Dynamics of fibrin formation within thrombi analyzed by change in fluorescent intensity following injury. (D) Representative images of platelet accumulation (green) and fibrin formation (red) in growing thrombi in cremaster arterioles in PPARα−/− mice treated with vehicle control (ctrl), DPAn-6, 11-HpDPAn-6, or 14-HpDPAn-6. Time after vascular injury is indicated above (n = 3 mice, 10-12 thrombi per mouse). (E) Dynamics of platelet accumulation in thrombi in PPARα−/− mice analyzed by change in fluorescent intensity. (F) Dynamics of fibrin formation in thrombi in PPARα−/− mice analyzed by change in fluorescent intensity following injury. (G) Mean tail bleeding of control mice (n = 24) or mice treated with DPAn-6 (n = 17) is denoted by the horizontal line. (H) The proposed model of DPAn-6 12-LOX oxylipins’ inhibitory effect on platelet activation. DPAn-6 12-LOX oxylipins derived from the elongation of DGLA into the bioactive lipids 11-HpDPAn-6 and 14-HpDPAn-6. DPAn-6 oxylipins regulate platelet function through activation of PPARα, which leads to inhibition of dense and α-granule release, calcium mobilization, and platelet reactivity in response to G protein–coupled receptor–based or immunoreceptor tyrosine-based activation motif–mediated platelet activation. Data are means ± SEM. ****P < .0001, 2-way ANOVA.
To determine the in vivo requirement for PPARα in the inhibition of platelet thrombosis by 11-HpDPAn-6 and 14-HpDPAn-6, thrombus formation was tested in PPARα−/− mice (Figure 7D). Thrombus formation was attenuated in PPARα−/− mice compared with WT mice (Figure 7E). However, treatment with DPAn-6, 11-HpDPAn-6, or 14-HpDPAn-6 failed to further inhibit thrombus formation in PPARα−/− mice (Figure 7D-E). Fibrin formation was significantly decreased in whole animal PPARα−/− mice compared with WT (Figure 7F). A difference in fibrin formation was observed following treatment with DPAn-6 vs 11-HpDPAn-6 or 14-HpDPAn-6. Although fibrin formation in PPARα−/− animals was not noticeably different with DPAn-6 treatment, it was absent in PPARα−/− animals treated with 11-HpDPAn-6 or 14-HpDPAn-6. To determine whether DPAn-6 or its 12-LOX oxylipins altered hemostasis, WT mice were given DPAn-6, 50 mg/kg per day, for 4 weeks. Tail bleeding time was assessed in these mice and showed no significant increase compared with control animals (Figure 7G).
Discussion
The current study elucidated for the first time the potential of DPAn-6 and its oxylipins to regulate platelet reactivity, clot formation, and thrombosis. Importantly, this study showed that DPA and its oxylipins regulate platelet function through a novel lipid signaling pathway involving activation of the nongenomic activities of PPARα. Although PPARα is predominantly known as a transcriptional regulator of genes involved in peroximal and mitochondrial β-oxidation and fatty acid transport, this study determined that the underlying mechanism by which the DPAn-6 oxylipins exert their antiplatelet effect is selectively through PPARα in a nontranscriptional manner. These observations were supported with the use of pharmacological and genetic models and confirmed with a transcriptional reporter assay. Both DPAn-6 oxylipins, 11-HpDPAn-6 and 14-HpDPAn-6, were observed to potently inhibit platelet activation (aggregation, calcium flux, PKC activation, and dense granule secretion), platelet accumulation in microfluidic perfusion chamber, and thrombus growth in vivo following vessel injury. Additionally, inhibition of fibrin formation by DPA oxylipins, especially 14-HpDPAn-6, was observed, indicating a possible inhibition of the coagulation pathway. Although the data presented here are highly supportive of the direct regulation of platelet function through activation of PPARα, other pathways may exist, such as secondary metabolism into dihydroxyeicosatetraenoic acid (diHETE) or trihydroxyeicosatetraenoic acid (triHETE) products through transcellular oxidation in the blood.29 These more complex oxidized metabolites may exert additional effects in vivo, as has been observed with DHA regulation of thrombus resolution through formation of proresolvins.35 Unexpectedly, a decrease in thrombosis was observed in PPARα−/− mice compared with WT mice in the in vivo response to vascular injury. Although PPARs are known to regulate a variety of cellular functions,36 their effect on thrombosis and coagulation in vivo is not well studied. Decreased thrombosis in PPARα−/− mice might be due to a combined effect of the PPAR on the platelet and endothelium. Hence, efforts are ongoing to create a platelet-specific PPARα−/− mouse that would enable future studies on the role of platelet-specific PPARα deficiency in the regulation of platelet function and thrombosis.
PPARα agonists, such as fenofibrate, are approved by the US Food and Drug Administration for secondary treatment of dyslipidemia and function to reduce triglycerides. Based on the PPARα binding data in Figure 4, 11-HpDPAn-6 and 14-HpDPAn-6 were observed to activate PPARα in the low nanomolar range and were 100- to 1000-fold more potent than the synthetic PPARα agonist WY-14643. Furthermore, pharmacological inhibition of PPARα, but not PPARβ or PPARγ, blocked the effects of oxylipin-mediated inhibition of platelet function. These observations suggest that 11-HpDPAn-6 and 14-HpDPAn-6 are potent and selective PPARα agonists and support that oxylipin-derived PPARα agonists may play a role in the prevention of dyslipidemia or thrombotic-related cardiovascular disease.
Although the role of PPARs has primarily been viewed as transcriptional regulation of gene expression, recent evidence presented here and elsewhere supports their ability to function in a nongenomic manner.34,37-39 PPARγ, for instance, has been demonstrated to play a role in the regulation of Syk and LAT and is activated by rosiglitazone,33 as well as by 15dPGJ2 and ciglitazone.34,39 Activation of PPARα with fenofibrate has been postulated to contribute to the repression of PKC activity.32 In contrast, although we observed inhibition of PKC activity by the DPAn-6 oxylipins, VASP phosphorylation was not observed at the protein kinase G site (S239) or protein kinase A site (S139), suggesting that regulation of platelets by DPAn-6 oxylipins is independent of cAMP or cyclic guanosine 3′,5′-cyclic monophosphate.
In vivo data presented here demonstrate that the vessels of mice administered DPAn-6 oxylipins are protected from vascular injury without exhibiting an increased bleeding risk. Further, the current work demonstrates that oxylipin-induced PPARα activation serves a regulated signaling function in at least a subset of cells expressing the PPARα protein, including platelets.32,40-42 Additionally, the data presented here elucidate a potential PPARα-dependent mechanism for regulation of platelet function induced by DPAn-6 12-LOX oxylipin products that includes inhibition of intraplatelet calcium mobilization and PKC activity, attenuation of secretion and aggregation, and inhibition of arterial thrombosis (Figure 7H). Hence, the PPARα noncanonical transcription-independent signaling mechanism explains in part how PUFA supplementation functions in the blood to attenuate or reduce thrombotic risk.
All oxylipins and protocols for isolating and/or synthesizing these oxylipins will be made available following e-mail requests to the corresponding author, Michael Holinstat (e-mail: mholinst@umich.edu).
Acknowledgments
The authors thank Josephine E. Lauderdale for contributions in assessing oxylipin binding to PPAR.
This work was supported, in part, by National Institutes of Health grants R01 HL114405 (M.H. and T.R.H.), R01 HL144660 (M.H.), and F31 HL129481 and F32 HL149280 (J.Y.) from the National Heart, Lung, and Blood Institute; R01 GM105671 (M.H. and T.R.H.), R35 GM131835 (M.H. and T.R.H.), and R01 GM1155884 (A.D.) from the National Institute of General Medicine; R03 DA042365 (A.D.) from the National Institute of Drug Abuse; and by American Heart Association Scientist Development Grant 15SDG25760064 (A.D.).
Authorship
Contribution: J.Y., T.R.H., and M.H. conceived the study; J.Y., A.Y., T.R.H., R.A., A.D., and M.H. designed the experiments; J.Y., A.Y., R.A., T.R.H., and M.H. wrote the manuscript; C.J.F., A.C., and A.D. critically read and edited the manuscript; and all authors performed experiments and analyzed the data.
Conflict-of-interest disclosure: M.H. is a consultant and equity holder in Veralox Therapeutics. T.R.H. is a member of the scientific advisory board for Veralox Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Michael Holinstat, University of Michigan Medical School, 1150 West Medical Center Dr, 2220D Medical Sciences Research Building III, Ann Arbor, MI 48109-5632; e-mail: mholinst@umich.edu.