Key Points
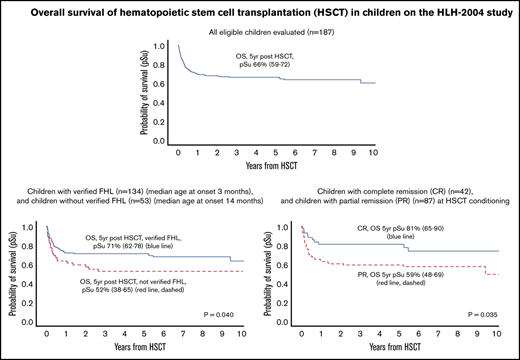
In 187 children with a transplant, 5-year OS post-HSCT was 66% in the entire cohort and 71% in children with FHL.
To spare children from unnecessary HSCT, pretransplant analyses, including possible confirmation of FHL, are recommended.

Abstract
We report the largest prospective study thus far on hematopoietic stem cell transplantation (HSCT) in hemophagocytic lymphohistiocytosis (HLH), a life-threatening hyperinflammatory syndrome comprising familial/genetic HLH (FHL) and secondary HLH. Although all patients with HLH typically need intensive anti-inflammatory therapy, patients with FHL also need HSCT to be cured. In the international HLH-2004 study, 187 children aged <18 years fulfilling the study inclusion criteria (5 of 8 diagnostic criteria, affected sibling, or molecular diagnosis in FHL-causative genes) underwent 209 transplants (2004-2012), defined as indicated in patients with familial/genetic, relapsing, or severe/persistent disease. Five-year overall survival (OS) post-HSCT was 66% (95% confidence interval [CI], 59-72); event-free survival (EFS) was 60% (95% CI, 52-67). Five-year OS was 81% (95% CI, 65-90) for children with a complete response and 59% (95% CI, 48-69) for those with a partial response (hazard ratio [HR], 2.12; 95% CI, 1.06-4.27; P = .035). For children with verified FHL (family history/genetically verified, n = 134), 5-year OS was 71% (95% CI, 62-78) and EFS was 62% (95% CI, 54-70); 5-year OS for children without verified FHL (n = 53) was significantly lower (52%; 95% CI, 38-65) (P = .040; HR, 1.69; 95% CI, 1.03-2.77); they were also significantly older. Notably, 20 (38%) of 53 patients without verified FHL had natural killer cell activity reported as normal at diagnosis, after 2 months, or at HSCT, suggestive of secondary HLH; and in addition 14 (26%) of these 53 children had no evidence of biallelic mutations despite having 3 or 4 FHL genes analyzed (natural killer cell activity not analyzed after 2 months or at HSCT). We conclude that post-HSCT survival in FHL remains suboptimal, and that the FHL diagnosis should be carefully investigated before HSCT. Pretransplant complete remission is beneficial but not mandatory to achieve post-HSCT survival. This trial was registered at www.clinicaltrials.gov as #NCT00426101.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a severe hyperinflammatory condition with an uncontrolled accumulation of macrophages and lymphocytes.1 HLH typically comes in a primary (genetic) form, which is almost always fatal, and a secondary (acquired) form. The most common form of primary HLH is familial HLH (FHL), which is autosomal recessive and caused by biallelic mutations in 1 of 4 genes (PRF1, UNC13D, STX11, and STXBP2), all associated with defective lymphocyte cytotoxicity.2-7 Primary HLH also includes other diseases such as X-linked lymphoproliferative disease (XLP), Griscelli syndrome type 2 (GS2), and Chédiak-Higashi syndrome (CHS). Secondary HLH (sHLH), on the contrary, is typically triggered by acquired conditions, the most common being severe infections, malignancies, and inflammatory disorders.3,8-10
Clinical manifestations of HLH include prolonged fever, splenomegaly, cytopenias, hypertriglyceridemia, hypofibrinogenemia, and hyperferritinemia.11-13 The most frequent severe sequelae in pediatric HLH are associated with central nervous system (CNS) involvement, often leading to neurologic deficits.1,14,15 Treatment of HLH aims to downregulate the hyperactive immune system; in addition, for primary HLH, hematopoietic stem cell transplantation (HSCT) is required for cure, as first shown by Fischer et al.16-18 HSCT is also needed for certain forms of sHLH, such as chronic active Epstein-Barr virus infection, and some cases of malignancy-associated HLH.19-21
As a result of collaborations worldwide and new treatment protocols, survival in HLH has improved dramatically.1,22-26 In 1994, the Histiocyte Society launched the first international therapeutic study on HLH, HLH-94, which recruited >200 eligible patients.27 It resulted in remarkably improved outcome, with a 5-year probability of overall survival (OS) of 54%.28 Transplant-related mortality (TRM) caused 26 of 31 deaths after HSCT in HLH-94 and is reportedly higher in HLH than in other nonmalignant conditions, with a large proportion of noninfectious pulmonary toxicity, infections, and sinusoidal obstruction syndrome/veno-occlusive disease.29-34
In HLH-2004, the second international HLH study launched by the Histiocyte Society, 5-year OS reached 61%, and pre-HSCT mortality was 19% compared with 26% in HLH-94 (using the same criteria).35 The current article summarizes the data on HSCT in children recruited to the HLH-2004 study; detailed pre-HSCT information is provided by Bergsten et al.35
Patients and methods
Patients
Data on HSCT from 187 eligible children (aged <18 years; fulfilled ≥5 of 8 diagnostic HLH-2004 criteria or had a molecular diagnosis consistent with FHL; no previous cytotoxic or cyclosporine treatment; and no known other underlying disease) were reported to the HLH-2004 study from 22 countries (Argentina, Austria, Bahrain, Brazil, Canada, Czech Republic, Denmark, Germany, Italy, Japan, Malaysia, The Netherlands, Norway, Oman, Portugal, Serbia, Spain, Sweden, Switzerland, Turkey, the United Kingdom, and the United States).12,35 In relation to our previous HLH-2004 report, follow-up data from 2 additional patients were available.35 Patient characteristics are detailed in Table 1.
A total of 134 patients were defined as having FHL if they had verified biallelic mutations in PRF1, UNC13D, STX11, or STXBP2 (n = 130)2,4-7 and/or a sibling with HLH (n = 34 [4 of whom were not analyzed for biallelic mutations]). FHL could not be verified in 53 of 187 eligible children (n = 24 aged <1 year and n = 29 aged ≥1 year at the start of HLH-2004 treatment). Of the 53 nonverified FHL patients, 15, 16, 6, and 13 patients were screened for 4, 3, 2, and 1 disease-causative genes, respectively. STXBP2 was identified first in 2009.2,6 As stated in the HLH-2004 protocol, children with other primary HLH diseases (GS2, n = 11; XLP1, n = 6; XLP2, n = 2; and CHS, n = 1) were evaluated separately.12
Patients underwent transplantation from 1 January 2004 to 31 December 2012; the last day of data entry was 31 December 2017. The Histiocyte Society, the ethics review board of Stockholm, Sweden, and the Swedish Medical Products Agency (EUDRACT 2005-003279-18) approved the HLH-2004 study, which was also registered at clinicaltrials.gov (#NCT00426101). Data were reported on clinical report forms associated with the study at transplantation, 100 days post-HSCT, and yearly thereafter. Reports on serious adverse events and mortality reports were sent to the principal investigator and were subsequently reviewed by the external Data Safety Monitoring Board.
Donor typing, conditioning, and graft-versus-host-disease prophylaxis
HLA typing results were reported for 6 loci, class I (A, B) and class II (DR). At first transplant, 44 patients (24%) had a matched-related donor (MRD), 60 (32%) had a matched unrelated donor (MUD), 11 (6%) had a haploidentical donor, 6 (3%) had a mismatched unrelated donor (MMUD), and 53 (28%) had umbilical cord blood (UCB) donor (no data, n = 13 [7%]) (Table 2).
According to the HLH-2004 treatment protocol, the modalities of the transplant procedure were decided by the transplantation team. A myeloablative conditioning regimen containing busulfan was suggested. Because only limited data on reduced-intensity conditioning were available at the time of writing the HLH-2004 protocol, it was not possible to make evidence-based suggestions on such regimens.12,36 In this report, in line with an article by Messina et al,32 the conditioning regimen when known was reported as busulfan based (n = 99) or treosulfan based (n = 20) if these drugs were included in the conditioning, independent of whether fludarabine was included, and fludarabine based if fludarabine but neither busulfan nor treosulfan was included (n = 39) (others, n = 3; no data, n = 26) (Table 2; supplemental Table 1). No information on the doses of each drug was reported in the clinical report forms.
As graft-versus-host disease (GVHD) prophylaxis, cyclosporine and methotrexate (MTX)/mycophenolate mofetil (MMF) were suggested in the HLH-2004 protocol. A total of 118 patients received cyclosporine; as monotherapy (n = 22), with short-course MTX (n = 52), MMF (n = 28), or prednisone/methylprednisolone (n = 24). Twenty-two received tacrolimus; as monotherapy (n = 4), with short-course MTX (n = 8), MMF (n = 5), or prednisone/methylprednisolone (n = 4). Some patients had multiple combinations; 35 had no data. In 10 patients, grafts were T-cell depleted.
HLH disease status at HSCT conditioning
HLH disease activity was assessed at the start of HSCT conditioning, analyzed both as single clinical parameters (using cutoff values or as continuous variables) or by combined parameters. Complete response was defined as no fever, no splenomegaly, absolute neutrophil count (ANC) ≥1.0 × 109/L, platelets ≥100 × 109/L, fibrinogen >1.5 g/L, and ferritin analyzed both for <500 µg/L or <2000 µg/L; one missing parameter (ie, 6 of 6 or 5 of 5 normal parameters) was allowed. Evaluations regarding the ferritin level of 2000 µg/L were post hoc analyses performed due to literature suggesting that this was a more appropriate cutoff value.37-39 Altogether, 131 patients (70%) had sufficient information for response evaluation (ie, ≥5 parameters evaluated). Partial response was defined as having 3 to 5 normal parameters of 6 measured, or 3 to 4 of 5 measured, in addition to 1 to 3 abnormal parameters. No response was defined as having >3 abnormal parameters.
Statistical methods
For comparison between groups, Fisher’s exact or Pearson’s χ2 test was used for categorical variables, and the Mann-Whitney U test was used for continuous variables. The distribution of continuous variables was assessed by using histograms and normal qq-plots. Ferritin, ANC, triglycerides, aspartate aminotransferase, alanine aminotransferase, and soluble CD25 were ln-transformed to limit the influence of outliers.
Kaplan-Meier estimates were used for OS and event-free survival (EFS) (calculated from the day of HSCT until last follow-up, where death, and death or second HSCT, were defined as events, respectively). Confidence intervals (CIs) (95%) for survival estimates were calculated by using the log-minus-log transformation.40 Cox proportional hazards models were used to calculate hazard ratios (HRs) with two-sided 95% CIs. We fitted adjusted Cox regression models for our main findings, adjusting for age at diagnosis and time to HSCT, respectively. Restricted cubic splines were used in Cox proportional hazards models to test the linearity assumption of the continuous variables listed in Table 1. No serious deviations from linearity were found. Cumulative incidence of TRM or death due to HLH relapse, or second HSCT, was compared between groups by using Gray’s test.41 P < .05 was considered significant. All analyses were performed for the entire cohort (N = 187), for children with verified FHL (n = 134), and for children without verified FHL (n = 53), separately. IBM SPSS for Windows (version 25; IBM SPSS Statistics, IBM Corporation), the function rcspline.plot in R package rms,42 or the function cuminc in R package cmprsk43 in R version 3.6.0 (R Core Team 2019) were used for the aforementioned analyses.44
Results
Patient characteristics
Of the187 eligible children, 134 had verified FHL; of the remaining 53 children, 24 were aged <1 year and 29 were aged ≥1 year at HLH-2004 treatment start (Table 1). The median age at the start of HLH-2004 treatment and at HSCT conditioning for the entire cohort was 138 and 371 days, respectively; 91 (49%) were aged <1 year at transplantation, and 21 (11%) were aged ≥6 years. The median time to transplantation was 148 days; 42% (n = 79), 64% (n = 120), and 86% (n = 160) underwent transplant within 4, 6, and 12 months from HLH-2004 therapy start.
Among the 134 children with verified FHL, 41 (31%) displayed biallelic mutations in PRF1, 66 (49%) in UNC13D, five (4%) in STX11, and 18 (13%) in STXBP2. Thirty-four of these 134 children (25%) had a sibling with HLH, of whom 4 were not analyzed for mutations. Seventy-eight (58%) of the verified FHL patients were aged <1 year at transplantation, and only 10 (8%) were aged ≥6 years. The median age was 103 days at the start of treatment and 309 days at HSCT. Median time to transplantation was 129 days; 48% (n = 64), 70% (n = 94), and 88% (n = 118) underwent transplant within 4, 6, and 12 months after the start of HLH-2004 therapy, respectively.
For children without verified FHL (n = 53), median time to transplantation (190 days) was significantly longer compared with children with verified FHL (P = .011), and about one-half of these children (n = 27 [52%]) had their transplant ≥6 months after the start of HLH-2004 therapy. These children were older than those with verified FHL; their median age was 441 days at the start of therapy (P < .001) and 651 days at HSCT (P < .001), and only 25% (n = 13) were aged <1 year at HSCT, whereas 21% (n = 11) were aged ≥6 years.
Survival
As of 31 December 2017, a total of 120 children (64%) were alive, with a follow-up of ≥3 years in 107, ≥4 years in 96, and ≥5 years in 81 from the first HSCT (supplemental Table 2). The 5-year OS post-HSCT was 66% (95% CI, 59-72) and EFS was 60% (95% CI, 52-67) (Figure 1A-B; Table 3). For children with verified FHL, 5-year OS was 71% (95% CI, 62-78) and EFS was 62% (95% CI, 54-70), whereas the 5-year OS for children without verified FHL was significantly lower (52%; 95% CI, 38-65) (P = .040; HR, 1.69; 95% CI, 1.03-2.77) and their EFS was nonsignificantly lower (52%; 95% CI, 38-65) (P = .27; HR, 1.32; 95% CI, 0.81-2.22) (Figure 1C-D). Nonverified FHL remained a statistically significant risk factor (P = .032; HR, 1.74; 95% CI, 1.05-2.88) for post-HSCT death when adjusting for time to HSCT but not when adjusting for age at diagnosis. For children with mutations in PRF1, UNC13D, STX11, and STXBP2, 5-year OS was 70% (95% CI, 64-82), 70% (95% CI, 54-79), 80% (95% CI, 20-97), and 71% (95% CI, 44-87), respectively, and OS in children with an affected sibling was 79% (95% CI, 62-90). OS in girls without verified FHL was 44% (95% CI, 24-63) compared with 75% (95% CI, 63-83) in those with verified FHL (P = .006), and for boys it was 61% (95% CI, 40-76) and 66% (95% CI, 52-76) (P = .90); the interaction analysis (nonverified FHL/verified FHL; female/male) yielded P = .062.
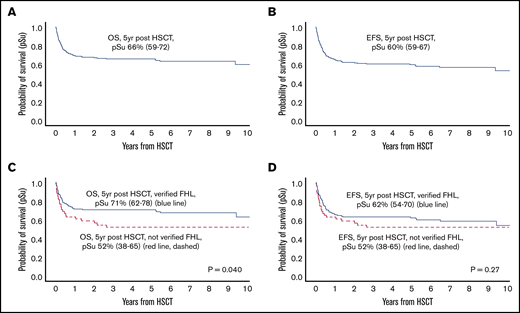
Kaplan-Meier estimates of survival after HSCT in the HLH-2004 study. Five-year probabilities of survival (pSu) are indicated with a 95% CI. OS or EFS is displayed, where death, and death or second HSCT, were defined as events, respectively. P values from Cox proportional hazards models. (A) OS for the entire HLH-2004 cohort (n = 187, events n = 67). (B) EFS for the entire HLH-2004 cohort (n = 187, events n = 78). (C) OS for children with verified FHL (n = 134, events n = 43, blue line) and children without verified FHL (n = 53, events n = 24, red line dashed); P = .040. (D) EFS for children with verified FHL (n = 134, events n = 54, blue line) and children without verified FHL (n = 53, events n = 24, red line, dashed); P = .27.
Kaplan-Meier estimates of survival after HSCT in the HLH-2004 study. Five-year probabilities of survival (pSu) are indicated with a 95% CI. OS or EFS is displayed, where death, and death or second HSCT, were defined as events, respectively. P values from Cox proportional hazards models. (A) OS for the entire HLH-2004 cohort (n = 187, events n = 67). (B) EFS for the entire HLH-2004 cohort (n = 187, events n = 78). (C) OS for children with verified FHL (n = 134, events n = 43, blue line) and children without verified FHL (n = 53, events n = 24, red line dashed); P = .040. (D) EFS for children with verified FHL (n = 134, events n = 54, blue line) and children without verified FHL (n = 53, events n = 24, red line, dashed); P = .27.
OS did not improve over time when comparing transplantation years 2004 to 2006, 2007 to 2009, and 2010 to 2012 in the entire cohort (P = .20). From 2010 to 2012, OS exceeded 80% (82%; 95% CI, 55-94) for FHL children with MUD (n = 17) (supplemental Table 3).
Natural killer cell activity in children without verified FHL undergoing transplant
Decreased natural killer (NK) cell activity at the onset of HLH is associated with deficient function of cytotoxic lymphocytes (typical for most patients with primary HLH) or with a low number of circulating cytotoxic cells (mostly seen in sHLH).45-47 To better understand if some of the children without verified FHL (n = 53) who underwent transplant could have possible sHLH, reported data on genetics and NK cell activity were reviewed in detail. Notably, 20 (38%) of 53 had NK cell activity reported normal at diagnosis, after 2 months, or at HSCT, which is suggestive of sHLH. Moreover, in addition 14 (26%) of these 53 additional children had no evidence of biallelic mutations despite having 3 or 4 FHL genes analyzed (as well as NK cell activity not analyzed after 2 months or at HSCT). Altogether, these 34 patients without evidence of primary HLH, who instead may have had sHLH, comprise 64% of this cohort of 53 patients (supplemental Table 4).
Notably, 11 (21%) of 53 children without verified FHL had low or absent NK cell activity after 2 months or at HSCT, possibly suggesting primary HLH. For 8 (15%) of 53 children, NK cell activity was not assessed/missing at 2 months or at HSCT, and mutation analysis was only performed for 2 (n = 1), 1 (n = 6), or no (n = 1) FHL genes; thus, for these children, the limited data cannot help in differentiating between primary vs secondary HLH.
Donor groups
The 5-year EFS for MRD, MUD, and UCB was 54% (95% CI, 38-67), 62% (95% CI, 48-73), and 58% (95% CI, 44-70), respectively, and the corresponding OS was 58% (95% CI, 42-71), 73% (95% CI, 60-83), and 60% (95% CI, 45-72) (Table 3; Figure 2A; supplemental Figure 1A). For children with verified FHL, the 5-year EFS was 58% (95% CI, 38-73), 65% (95% CI, 48-77), and 66% (95% CI, 49-79) for MRD, MUD and UCB. The corresponding 5-year OS was 64% (95% CI, 45-78), 78% (95% CI, 63-88), and 69% (95% CI, 52-81) (Table 3; Figure 2B; supplemental Figure 1B).
![Kaplan-Meier estimates of EFS based on HSCT donor and conditioning in the HLH-2004 study. Five-year probabilities of survival (pSu) are indicated with a 95% CI. Death or second HSCT were defined as events. P values from Cox proportional hazards models. (A) EFS for the entire cohort based on donor: MRD (n = 44, event n = 21, blue line); MUD (n = 60, event n = 24, red line, dashed); UCB (n = 53, event n = 23, black line, dotted)], P = .52. [MUD vs MRD, P = .26; UCB vs MRD, P = .61]. (B) EFS for children with verified FHL based on donor (MRD, n = 31, event n = 14, blue line; MUD, n = 45, event n = 18, red line, dashed; UCB, n = 39, event n = 14, black line, dotted; P = .65 (MUD vs MRD, P = .40; UCB vs MRD, P = .44). (C) EFS for the entire cohort based on conditioning (busulfan-based, n = 99, events n = 42, blue line; fludarabine based, n = 39, events n = 18, red line dashed; treosulfan based, n = 20, events n = 5, black line, dotted; P = .31 (fludarabine vs busulfan, P = .74; treosulfan vs busulfan, P = .16). (D) EFS for children with verified FHL based on conditioning (busulfan based, n = 69, events n = 27, blue line; fludarabine based, n = 26, events n = 12, red line, dashed; treosulfan based, n = 18, events n = 5, black line, dotted; P = .49 (fludarabine vs busulfan, P = .49; treosulfan vs busulfan, P = .43).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/15/10.1182_bloodadvances.2020002101/4/m_advancesadv2020002101f2.png?Expires=1763478924&Signature=4GMBh9CqTDB0v8RVNwPjMRnri0kxOLLOjXx2hIJ5oS-m3byMXiWMeDqBiyThsRacAiKnsNau~ebiHTBP0zQ6SVIuLoYt8WR8EFOpNp8cVuC7nRSxQYWxEN4Lv53qgPyUybujVRxiVaoj~EaZ8sHKlZhOwzA3KTuofTbqgmKQCFKuBE6DIY2wUROcooVfIcibnhjD9etFaxsou~9ZE7e76CwueCoGvZEJk8ppWJcTBSG~HW9yZO9o-CAhHIQ2tct6YC-sz6O4M-4g9rWIsO0y7ldj0MOO~qnkzrzfOo9MIwtotBe60gk5ehavo3kPCM52XqDwkfBzOUa8g1LV~WrIUA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Kaplan-Meier estimates of EFS based on HSCT donor and conditioning in the HLH-2004 study. Five-year probabilities of survival (pSu) are indicated with a 95% CI. Death or second HSCT were defined as events. P values from Cox proportional hazards models. (A) EFS for the entire cohort based on donor: MRD (n = 44, event n = 21, blue line); MUD (n = 60, event n = 24, red line, dashed); UCB (n = 53, event n = 23, black line, dotted)], P = .52. [MUD vs MRD, P = .26; UCB vs MRD, P = .61]. (B) EFS for children with verified FHL based on donor (MRD, n = 31, event n = 14, blue line; MUD, n = 45, event n = 18, red line, dashed; UCB, n = 39, event n = 14, black line, dotted; P = .65 (MUD vs MRD, P = .40; UCB vs MRD, P = .44). (C) EFS for the entire cohort based on conditioning (busulfan-based, n = 99, events n = 42, blue line; fludarabine based, n = 39, events n = 18, red line dashed; treosulfan based, n = 20, events n = 5, black line, dotted; P = .31 (fludarabine vs busulfan, P = .74; treosulfan vs busulfan, P = .16). (D) EFS for children with verified FHL based on conditioning (busulfan based, n = 69, events n = 27, blue line; fludarabine based, n = 26, events n = 12, red line, dashed; treosulfan based, n = 18, events n = 5, black line, dotted; P = .49 (fludarabine vs busulfan, P = .49; treosulfan vs busulfan, P = .43).
Kaplan-Meier estimates of EFS based on HSCT donor and conditioning in the HLH-2004 study. Five-year probabilities of survival (pSu) are indicated with a 95% CI. Death or second HSCT were defined as events. P values from Cox proportional hazards models. (A) EFS for the entire cohort based on donor: MRD (n = 44, event n = 21, blue line); MUD (n = 60, event n = 24, red line, dashed); UCB (n = 53, event n = 23, black line, dotted)], P = .52. [MUD vs MRD, P = .26; UCB vs MRD, P = .61]. (B) EFS for children with verified FHL based on donor (MRD, n = 31, event n = 14, blue line; MUD, n = 45, event n = 18, red line, dashed; UCB, n = 39, event n = 14, black line, dotted; P = .65 (MUD vs MRD, P = .40; UCB vs MRD, P = .44). (C) EFS for the entire cohort based on conditioning (busulfan-based, n = 99, events n = 42, blue line; fludarabine based, n = 39, events n = 18, red line dashed; treosulfan based, n = 20, events n = 5, black line, dotted; P = .31 (fludarabine vs busulfan, P = .74; treosulfan vs busulfan, P = .16). (D) EFS for children with verified FHL based on conditioning (busulfan based, n = 69, events n = 27, blue line; fludarabine based, n = 26, events n = 12, red line, dashed; treosulfan based, n = 18, events n = 5, black line, dotted; P = .49 (fludarabine vs busulfan, P = .49; treosulfan vs busulfan, P = .43).
Eleven children had a haploidentical donor (EFS, 82%; 95% CI, 45-95), all from a few transplantation centers, and six had an MMUD (EFS, 67%; 95% CI, 20-90). Neither EFS nor OS was significantly influenced by the choice of donor (Figure 2A-2B; supplemental Figure 1).
Conditioning, engraftment, GVHD, and chimerism
The choice of conditioning regimen did not statistically significantly affect the 5-year EFS for all children (busulfan based, 59% [95% CI, 48-68]; fludarabine based, 50% [95% CI, 32-66]; treosulfan based, 80% [95% CI, 55-92]). The corresponding 5-year OS values were 63% (95% CI, 53-72), 69% (95% CI, 51-81), and 80% (95% CI, 55-92) (Table 3; Figure 2C; supplemental Figure 2A). In neither of the subgroups (with or without verified FHL) did conditioning statistically significantly influence EFS or OS (Table 3; Figure 2D; supplemental Figure 2B-D).
Information on engraftment by day 100 was available for 150 children (no data, n = 37), of whom 137 (91%) were reported to have engraftment (100 of 107 [93%] for children with verified FHL and 37 of 43 [86%] for children without FHL). Thirty-one (22%) of 138 had acute GVHD grade II to IV reported at 100 days, and 21 had chronic GVHD (years 1-5 post-HSCT). Complete chimerism at day 100 was reported in 72 children (39%), and mixed chimerism was reported in 47 (25%) (no data, n = 68).
Disease activity at conditioning
Clinical and laboratory parameters were evaluated as single parameters at time of HSCT conditioning, either with normal cutoff values or as continuous variables. Using cutoff values, no statistically significant differences regarding survival were found. Of children with verified FHL, 27 (28%) had ANC <1.0 × 109, and 17 (85%) had low/absent NK cell activity, compared with 3 (15%) and 5 (50%), respectively, of children aged ≥1 year without verified FHL (Table 1). One-fifth (n = 30) of all patients had neurologic alterations at HSCT conditioning, and 24 (44%) displayed pathologic cerebrospinal fluid cells and/or protein.
When using continuous variables, a statistical significance in OS was found for ferritin (ln-transformed) both in the entire cohort (HR for a 1-unit increase in the natural logarithm of ferritin (µg/L) = 1.23; 95% CI, 1.03-1.48; P = .023) and in the children with verified FHL (HR, 1.29; 95% CI, 1.01-1.66; P = .040) (Table 4). In the FHL cohort, the difference remained statistically significant when adjusting for age at diagnosis and time to HSCT, both separately and in the same model.
With regard to response at HSCT, the 5-year OS for all children with complete response (using ferritin <500 µg/L) was 81% (95% CI, 65-90) and 59% (95% CI, 48-69) for those with partial response (HR, 2.12; 95% CI, 1.06-4.27; P = .035) (Tables 3 and 4; Figure 3A). Partial response compared with complete response remained statistically significant for post-HSCT death (HR, 2.03; 95% CI, 1.01-4.11; P = .048) when adjusting for age at diagnosis and time to HSCT. By using ferritin levels <2000 µg/L as the cutoff, 5-year OS for all children with complete remission was 76% (95% CI, 62-85) and 61% (95% CI, 49-71) with partial response (HR, 1.74; 95% CI, 0.94-3.22; P = .080) (Tables 3 and 4; Figure 3B).

Kaplan-Meier estimates of OS based on complete or partial response at HSCT in the HLH-2004 study. Displayed are OS where death was defined as event. Five-year probabilities of survival (pSu) are indicated with a 95% CI. P values from Cox proportional hazards models. Complete response (CR) was defined as no fever, no splenomegaly, ANC ≥1.0 × 109/L, platelets ≥100 × 109/L, fibrinogen >1.5 g/L, and a ferritin cutoff of either <500 µg/L or <2000 µg/L (allowing missing information on one of the these parameters). Partial response (PR) was defined as having 3 to 5 normal parameters. (A) OS for the entire cohort based on CR (n = 42, events n = 10, blue line) or PR (n = 87, events n = 37, red line dashed); P = .035, using the ferritin cutoff of <500 µg/L. (B) OS for the entire cohort based on CR (n = 55, events n = 15, blue line) or PR (n = 76, events n = 31, red line dashed); P = .080, using the ferritin cutoff of <2000 µg/L.
Kaplan-Meier estimates of OS based on complete or partial response at HSCT in the HLH-2004 study. Displayed are OS where death was defined as event. Five-year probabilities of survival (pSu) are indicated with a 95% CI. P values from Cox proportional hazards models. Complete response (CR) was defined as no fever, no splenomegaly, ANC ≥1.0 × 109/L, platelets ≥100 × 109/L, fibrinogen >1.5 g/L, and a ferritin cutoff of either <500 µg/L or <2000 µg/L (allowing missing information on one of the these parameters). Partial response (PR) was defined as having 3 to 5 normal parameters. (A) OS for the entire cohort based on CR (n = 42, events n = 10, blue line) or PR (n = 87, events n = 37, red line dashed); P = .035, using the ferritin cutoff of <500 µg/L. (B) OS for the entire cohort based on CR (n = 55, events n = 15, blue line) or PR (n = 76, events n = 31, red line dashed); P = .080, using the ferritin cutoff of <2000 µg/L.
Second transplantation
Twenty children (18 with verified FHL) had a second HSCT, 4 within 100 days from the first HSCT and altogether 12 during the first year (missing data, n = 3). Donors at first HSCT were MUD (n = 8), UCB (n = 6), MRD (n = 4), haploidentical (n = 1), and MMUD (n = 1). Eight children had another donor for the second HSCT, whereas 5 children had the same (missing information, n = 7). Conditioning for the first HSCT was busulfan based (n = 9), fludarabine based (n = 8), and treosulfan based (n = 1) (missing information, n = 2). Notably, the 5-year cumulative incidence of a second transplantation was 23% (95% CI, 12-43) after fludarabine-based conditioning and 8% (95% CI, 4-14) after busulfan/treosulfan-based conditioning (P = .029).
Ten of 20 children were alive after the second HSCT, all with >3 years of follow-up after the second HSCT, whereas 7 (MRD, n = 1; MUD, n = 1; UCB, n = 5) had died (exact duration of follow-up is missing for 3 additional survivors). Two children had a third HSCT, and both were alive at last follow-up.
Mortality
Of the deceased children (n = 67 of 187 [36%]), 36 (54%) died within 100 days and 58 (87%) died during the first year post-HSCT. Nine died ≥1 year post-HSCT (six during years 2 and 3, and three after year 5). These 67 children included 43 (32%) of 134 with verified FHL and 24 (45%) of 53 without verified FHL. Thirty-six children (54%) were reported to have died of TRM (including five after their second HSCT), 18 (27%) due to HLH relapse (nine <100 days after HSCT; six with nonverified FHL, and three with FHL [one after second HSCT]). The cause of death (TRM or HLH relapse) could not be determined in 5 children, was reported as other causes in 4, and information was missing in 4.
Children without verified FHL were more likely to die of HLH relapse than children with verified FHL. Their 5-year cumulative incidence of HLH relapse death was 18% (95% CI, 10% to 32%) compared with 6% (95% CI, 3% to 12%) for children with verified FHL (P = .0123). Nine children without verified FHL died of HLH relapse, 8 aged ≥1 year at the start of HLH-2004 treatment and 4 aged >6 years. In children with verified FHL, all 9 who died of HLH relapse were aged <1 year at treatment start.
The causes of death are detailed in supplemental Table 5. About one-half of the deaths within 100 days were attributed to treatment-related lung and liver complications. One 17-year-old boy died of acute myeloid leukemia 2 years after start of HLH-2004 treatment, but etoposide was only administered for 3 weeks due to pneumonia and bacteremia, after which he was in remission.
Outcome of other primary HLH patients
In addition to the 134 children with verified FHL included in the study, 20 children with primary HLH underwent HSCT following HLH-2004 treatment (XLP, n = 8; GS2, n = 11; CHS, n = 1). Donors used were MRD (n = 5), MUD (n = 9), haploidentical (n = 3), MMUD (n = 1), and UCB (n = 1) (no data, n = 1). The conditioning regimens were busulfan based (n = 8), fludarabine based (n = 8), and treosulfan based (n = 1) (no data, n = 3). At last follow-up, 14 of 20 children were alive (5 of 6 children with XLP1, 1 of 2 with XLP2, 7 of 11 with GS2 and the 1 child with CHS), with a 5-year OS of 70% (95% CI, 45-85) and a 5-year EFS of 70% (95% CI, 45-85).
Discussion
We report the largest prospective study on HSCT for HLH thus far, comprising 187 children receiving 209 transplantations within the HLH-2004 study conducted with wide international collaboration. Survival in HLH improved dramatically worldwide after the introduction of the HLH-94 treatment protocol.28 In the subsequent study (HLH-2004), 5-year OS reached 61%, and pre-HSCT mortality was reduced from 26% in HLH-94 to 19% (using the same criteria).35 However, the overall 5-year OS post-HSCT of 66% (71% in children with verified FHL) in HLH-2004 reported here was not better than the 64% reported in HLH-94.31 In both protocols, myeloablative conditioning containing busulfan was suggested, which may in part explain the similar results. Nevertheless, HLH-2004 data could help to understand how to improve HSCT survival in HLH.
Notably, 53 (28%) of 187 children who underwent transplant had no family history, cytotoxicity data, or biallelic mutations suggesting verified FHL. Because not all children were analyzed for all FHL-related genes, some children reported as “without verified FHL” may still have had FHL. Remarkably, the age at start of HLH-2004 treatment was significantly higher in children without verified FHL compared with those with FHL (median, 441 days vs 103 days, respectively; P < .001); their median time to HSCT was significantly longer (P = .011), and, moreover, their 5-year OS was significantly lower (52% vs 71%; P = .040). Did these children really have FHL? Indeed, 34 (64%) of these 53 children had either no evidence of defective NK cell activity (n = 20) or, in patients for whom NK-cell activity was not analyzed after 2 months or at HSCT, no biallelic mutations in 3 or 4 FHL-related genes analyzed (n = 14). This suggests that a considerable proportion of the children undergoing transplant may actually not have had FHL, although we cannot exclude other relevant indications to transplant. Possible alternative diagnoses include various forms of sHLH, such as undetected malignancies (malignancy-associated HLH) as well as infection-associated HLH and MAS-HLH (eg, reactivating autoimmune/autoinflammatory diseases), particularly in children aged ≥1 year at the start of HLH-2004 (n = 29; median starting age, 1360 days).21 Similarly, children without verified FHL died of HLH relapse significantly more often than children with verified FHL (P = .012). With the aim to refine the diagnosis of FHL, because it is known that NK cell activity can be transiently reduced during active sHLH,46,47 we suggest retesting cytotoxicity function (including CD107a mobilization, perforin expression, and SAP/XIAP expression) pretransplant at a time of no HLH disease activity in patients lacking evidence of biallelic FHL-causing mutations.48-50
Timing of HSCT may be critical in FHL, also because prolonged disease activity increases the risk of irreversible CNS damage, which is the most important long-term complication in FHL.15 Disease control achieved by initial treatment is associated with a better outcome, as illustrated according to ferritin level: a one unit increase in the natural logarithm was associated with significantly increased risk of death in all patients (HR, 1.23; P = .023) as well as in verified FHL patients (HR, 1.29; P = .040). Notably, although the 5-year OS for all children with complete response (using a ferritin cutoff of <500 µg/L) at HSCT conditioning was better (81%) than for those with partial response (59%; P = .035), partial remission should not preclude performing HSCT in FHL.31,32,51 Therefore, children for whom nonactive disease is difficult to achieve (particularly in those with CNS involvement) would likely benefit from prompt HSCT as previously suggested.
The choice of HSCT donor may be crucial. The 78% 5-year OS in FHL patients with MUD compared favorably with the 64% outcome with MRD (P = .21), thus confirming that the use of MUD is a safe and valuable option (Table 4; supplemental Figure 1B). Whether the nonsignificantly inferior outcome associated with MRD represents a true phenomenon remains to be determined.
Is the choice of the conditioning regimen related to outcome? The 5-year OS for treosulfan-, fludarabine-, and busulfan-based regimens in children with verified FHL was 78%, 76%, and 70%, respectively. Remarkably, rescue transplantations may be beneficial in selected patients, as 13 (65%) of 20 patients with a second transplant and both patients with a third transplant were alive. Not surprisingly, second transplants were significantly more frequent after fludarabine-based conditioning than after busulfan-based/treosulfan-based conditioning (P = .029). Conversely, fludarabine-based reduced-intensity condition reportedly has less TRM than traditional busulfan-based myeloablative conditioning, albeit a higher degree of mixed chimerism, secondary graft failure, and relapse rates.32,36,52-57
This study has limitations, including the rate of missing values, likely due to its multicenter design. Another limitation is that, due to the year of writing the protocol, information on only 6 HLA loci for donor typing were requested. Because only 1 child was aged <3 months at the time of transplant, the study cannot advise on HSCT in such very young patients. Notably, results of this pediatric cohort cannot be fully translated to adult patients with HLH, because underlying diseases, as well as induction and conditioning therapy, differ significantly.21
We conclude that: (1) patients with nonverified FHL (possibly including sHLH cases) seem to do worse than verified FHL patients; (2) a thorough patient selection with pretransplant analyses, including confirmation of FHL, is recommended; and (3) pretransplant complete remission is beneficial but not mandatory to achieve post-HSCT survival. Finally, we are optimistic that post-HSCT survival in FHL can be improved based on these lessons, on rapid availability of functional and genetic diagnostic tests leading to earlier transplants, on promising studies of treosulfan-based conditioning that reportedly have less extramedullary toxicity than other myeloablative conditioning regimens,52,54 on targeted submyeloablative busulfan administration,58 and on more experience with fludarabine-based reduced-intensity condition.36,53,55-59
Requests for data may be submitted to the corresponding author (Jan-Inge Henter; e-mail: jan-inge.henter@ki.se).
Acknowledgments
The authors are grateful to all reporting clinicians, and they thank previous data managers Martina Löfstedt, Désirée Gavhed, and Tatiana von Bahr Greenwood for their contribution to the study registry. They also thank their HLH-2004 collaborators Jorge Braier, Maarten Egeler, Lisa Filipovich, and Shinsaku Imashuku for valuable contributions.
The work was supported by grants from the Swedish Childhood Cancer Foundation (KP2018-0005), the Swedish Research Council (2011-3897), the Cancer and Allergy Foundation of Sweden (No. 280), Stockholm Country Council (ALF-project) (SLL20180318), the Italian Ministry of Health (Ricerca finalizzata 2004 and Ricerca finalizzata TOS-2008-1219488), and the German Childhood Cancer Foundation (DKS 2016.04 and DKS 2018.11).
Authorship
Contribution: J.-I.H., A.H., M.A., G.J., S.L., and K.L.M. planned the study, with J.-I.H. as principal investigator and A.H. as study coordinator; J.-I.H., M.A., I.A., E.I., G.J., K.L., K.L.M., M.M., V.N., D.A.R., and E.S. recruited patients; I.H.M. provided statistical advice and analyses; J.W. served as HSCT advisor; E.B. performed data entry, compiled data, and performed statistical analyses; E.B., A.H., and J.-I.H. analyzed results; and E.B. and J.-I.H. drafted the manuscript, which was reviewed and approved by all authors.
Conflict-of-interest disclosure: I.A., A.H., K.L., K.L.M., E.S., and J.-I.H. serve as consultants for SOBI. A.H. serves as a speaker for Novartis. The remaining authors declare no competing financial interests.
Correspondence: Jan-Inge Henter, Childhood Cancer Research Unit, Karolinska Institute, Tomtebodavägen 18A, SE-171 77 Stockholm, Sweden; e-mail: jan-inge.henter@ki.se.