Abstract
Sickle cell disease (SCD) places a heavy burden on a global and increasing population predominantly resident in resource-poor and developing countries. Progress continues to be made in preventing childhood mortality, and increasing numbers of chronically ill adults with disease are requiring care for disease sequelae. Curative therapies for SCD are therefore attractive to physicians and investigators focused on SCD. Gene therapies are being developed, and several are now in various stages of early-phase human clinical trials. However, we must also pursue avenues through which we can do the most good for the most people alive today. Such efforts include improving our understanding of disease mechanisms and which disease sequelae most strongly affect survival and interfere with quality of life. The pathways leading to disease sequelae are multiple, complex, and highly interactive. Four drugs have now been approved by the US Food and Drug Administration for SCD; however, each has a distinct mechanism and a measurable but limited effect on the many clinical sequelae of SCD. We therefore need to learn how to approach multi-agent therapy for SCD. The order of addition of each agent to treat a specific patient will need to be guided by response to previous therapy, risk factors identified for specific disease outcomes, and clinical studies to determine more comprehensively how the 4 currently approved drugs might interact and produce (or not) additive effects. Moreover, this will have to be accomplished with defined end points in mind, according to which pose the greatest threats to quality of life as well as survival.
Where we are
Sickle cell disease (SCD) places a heavy burden on an increasingly widespread population throughout the world. Although only ∼100 000 to 120 000 of the ∼330 million people in the United States (0.036%) live with SCD,1 ∼20 million people are affected by SCD worldwide. Globally, ∼312 000 children are born with SCD each year.2 Most of the people affected by SCD live in developing countries with scarce resources to devote to health care. Thus, although survival is improving in India and in African countries, and adults with SCD are no longer highly unusual in those settings, the average survival with SCD still means that death during childhood is far more likely than survival to adulthood, with mortality under the age of 5 years estimated to be 50% to 90% in low-income countries.3 In addition, as we have learned in more resource-rich countries, survival to adulthood results in a high burden of disease-related complications during adult life, with multiple types of end-organ damage causing both shortened survival as well as substantially impaired quality of life. In addition, although death from SCD during childhood is relatively rare (<4%) in the United States,4 the nation spends approximately $1 billion annually on care for individuals with SCD.5
Curative therapies for SCD are therefore attractive to physicians and investigators focused on SCD, although such therapy offers both potential benefits and risks to patients. The first curative therapy to arrive on the horizon was hematopoietic stem cell transplant (HSCT). However, it became clear early on that this procedure was extremely challenging when performed in patients with SCD. Initial success was observed in young children, whereas success in older children and adults came at the price of a great deal of experimentation and high mortality rates during the early years of this effort. Although we now can perform HSCT for both children and adults with increasing success,6,7 HSCT has thus far reached only ∼2000 individuals worldwide, with overall survival of ∼95% and an average age at HSCT of 10 years.8 Thus, under the best circumstances, for the next few decades, HSCT will likely remain available to only a minority of patients due to donor availability and high resource requirements, although progress is being made in utilization of alternative donors, such as haploidentical family members.9
Meanwhile, gene therapies are being developed, and several are now in various stages of early-phase human clinical trials. Countries with robust medical research enterprises, including the United States, are increasingly focusing on gene therapy for hemoglobinopathies.10-14
Generally, gene therapy may take a variety of approaches, including: (1) addition of a helpful gene; (2) gene knockdown (eg, Bcl11A) to improve the phenotype; (3) direct globin gene editing to “correct” the mutation present (eg, changing a hemoglobin S [HbS]–encoding gene to one encoding hemoglobin A); and (4) gene editing of globin regulatory elements, to at least partially reverse the normal hemoglobin switching from fetal to adult hemoglobin. The gene addition approach may entail adding a gene encoding γ globin, a β/γ hybrid, an anti–sickling β-globin [eg, βA(T87Q)], or simply β-globin.15 Nonetheless, although gene therapy is improving relatively fast, there is still a long way to go. Continuous iterative improvement remains needed in every aspect of gene therapy development, including: (1) quality of viral delivery systems; (2) quality and quantity of hematopoietic stem cells harvested; (3) optimization of the gene modification system in hematopoietic stem cells; (4) choice of recombination pathway (homologous vs non-homologous); (5) identification of the best gene targets; (6) cell manufacturing; (7) preparation regimens to allow the bone marrow to receive genetically modified cells with minimized toxicity; (8) problems related to off-target effects; (9) optimization of preclinical models for testing of developing gene therapy strategies; and (10) parameters that should be used to define cure.
Given the current infrastructure present where most SCD patients are located, and to reach a significant fraction of people living with SCD today, gene therapy will also need to involve minimal bone marrow ablation and in vivo transduction (ie, Star Trek medicine). As in the Starship Enterprise sickbay, the patient would ideally be successfully treated by one injection of a healing factor and require no further care. Alternatively, if we will not be able to offer curative therapies to the vast majority of people currently living with SCD during their lifetimes, we must provide those patients alive today with alternative therapies to improve survival and quality of life.
Realizing the difficulties confronting gene therapy at this time, pharmaceutical companies and investigators have also been trying to develop pharmacologic methods to affect the genes controlling hemoglobin switching and thus increase fetal hemoglobin (HbF) expression. At least 2 loci independently and strongly affect HbF (γ/δ globin) suppression: Bcl11A and LRF/ZBTB7A. Potentially therapeutic small molecules have reportedly been identified, and early-phase clinical trials are in the planning stages to determine both their safety and whether they can indeed reactivate HbF expression in vivo.16 Other molecules may bind to and affect the globin control region, facilitating expression of γ globin.17 Although not curative per se, such therapeutic agents might be able to accomplish what is targeted by many gene therapies; that is, using increased HbF expression to improve cell biology and forestall the damage caused by sickle red cell sickling, hemolysis, and abnormal circulatory behavior.
The root of the problem
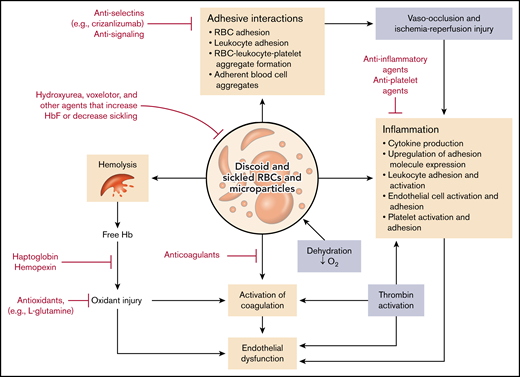
Of course, the root of the problem in SCD is the sickle red blood cell (SS RBC), a red blood cell containing predominantly HbS or HbS and another abnormal hemoglobin that participates in hemoglobin polymerization in the setting of hemoglobin deoxygenation. HbS is an inherently unstable hemoglobin in the circulation; it undergoes deoxygenation under higher oxygen tension than normal hemoglobin, forms polymers when deoxygenated, and ultimately leads to deformation of red blood cells (sickling) and hemolysis. However, exacerbating this process is a panoply of additional effects arising from perturbation of the RBCs by HbS (Figure 1).18 These effects include but are not limited to the following pathophysiologic processes, as partly illustrated in Figure 1:
Abnormal cation content and dehydration,19 leading to increased cellular viscosity;
Surface extracellular phosphatidylserine exposure, leading to activation of coagulation pathways20-22 ;
Metabolic abnormalities, including adenosine triphosphate and glutathione deficiency and high 2,3-diphosphoglycerate23-25 ;
Oxidative damage to membranes, accompanied by membrane instability and abnormal deformability and rheology26,27 ;
Abnormally high intracellular signaling activity, likely due to young RBC age and altered gene expression in stress erythropoiesis27-32 ;
Abnormally increased adhesive properties33-35 ;
Abnormal cell–cell signaling, leading to activation of leukocytes,36 endothelial cells,37 and probably platelets; and
Deficient expression of antioxidant compounds due to alteration in gene regulation and metabolism.32
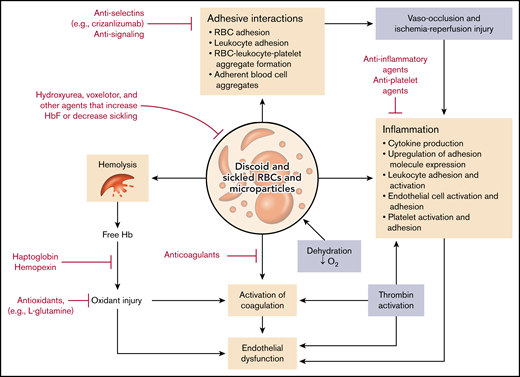
Pathophysiologic pathways and drug interventions in SCD. The pathologic RBCs containing HbS and microparticles derived from them give rise to a multiplicity of highly interactive pathways that contribute to hemolysis and organ damage. Various FDA-approved drugs as well as other classes of compounds may serve to reduce one or more of these processes and thus ameliorate the symptoms of the disease. O2, oxygen.
Pathophysiologic pathways and drug interventions in SCD. The pathologic RBCs containing HbS and microparticles derived from them give rise to a multiplicity of highly interactive pathways that contribute to hemolysis and organ damage. Various FDA-approved drugs as well as other classes of compounds may serve to reduce one or more of these processes and thus ameliorate the symptoms of the disease. O2, oxygen.
Thus, the SS RBC stands at the center of the vicious cycle of SCD that gives rise to hemolysis and anemia; vaso-occlusion with attendant pain, acute organ damage, and vasculopathy; and chronic tissue damage and vasculopathy leading to irreversible end-organ dysfunction. Importantly, the pathways leading to these downstream sequelae are multiple, complex, and highly interactive.
Doing the most good for the most people
Given our limited ability to affect the genetic root cause of SCD at this time, how can we do the most good for the most people alive today? To improve survival, we first must answer a critical question: Which sequelae are most associated with mortality? The answers, however, likely depend on geography and patient age. Although childhood mortality is low in the United States and countries with similar resources, it is common in Africa and India. Causes of disability also vary by both age and geography.
SCD results in multiple varieties of acute and chronic end-organ damage. These comprise cardiovascular and cardiopulmonary effects, including congestive heart failure and pulmonary hypertension; pulmonary effects, including acute chest syndrome; musculoskeletal effects, including dactylitis and avascular necrosis; neurologic effects, including both hemorrhagic and infarctive strokes, seizure disorders, and cognitive impairment; renal effects, including hyposthenuria, proteinuria, and end-stage renal disease; splenic sequelae, including splenic infarcts, splenic sequestration, and immunologic deficiencies associated with asplenia; hepatobiliary effects, including cholelithiasis and hepatic sequestration; and priapism. Among these sequelae, the ones most associated with mortality are acute chest syndrome,38 central nervous system effects (both strokes and presence of a seizure disorder, presumably reflecting silent infarcts),39-41 sickle cell nephropathy, as identified by the presence of macroalbuminuria,39,42 and pulmonary hypertension, as defined by elevated tricuspid regurgitant jet velocity. In addition, however, the frequency of vaso-occlusive (VOC) episodes is a predictor of mortality.43,44 Thus, therapeutic maneuvers that address these sequelae of SCD should extend survival. However, at the same time, we must remember that patients do not only want to live longer, they want to live better lives.
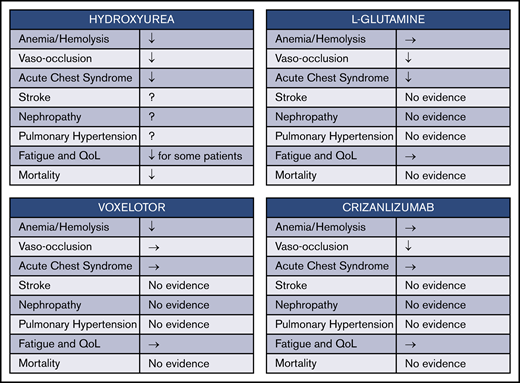
Four drugs have now been approved by the US Food and Drug Administration (FDA) for treatment of SCD. In reviewing the benefits of their use (Figure 2), however, we see that there is a long way to go to achieve both symptom relief and improved survival. Hydroxyurea seems to ameliorate many aspects of SCD, including anemia, VOC frequency, and acute chest syndrome frequency, while also improving overall survival. However, hydroxyurea is only partially effective in preventing stroke and may not have a major effect on the development of nephropathy and pulmonary hypertension, although definitive evidence is lacking. The 3 other more recently approved drugs for SCD are all individually somewhat less broad in their effects. l-glutamine decreased VOC frequency by ∼25%, while also reducing the frequency of acute chest syndrome significantly and hospitalization frequency by about one-third.45 However, the study leading to FDA approval failed to show any change in hematologic parameters such as anemia, and there was also no effect documented for well-being and quality of life measures. In addition, the mechanism whereby l-glutamine exerts its effects remains uncharacterized. Voxelotor, which binds to and shifts the oxygen dissociation curve of HbS and thereby inhibits deoxygenation and sickling, seems to substantially reduce hemolysis and ameliorate anemia in most patients with SCD.46,47 However, the study leading to its approval by the FDA for SCD treatment did not show any salutatory effects on VOC or acute chest syndrome frequency,47 nor is there evidence that the drug might be effective prophylaxis for stroke, nephropathy, or pulmonary hypertension. Crizanlizumab, which is a humanized monoclonal antibody directed against P-selectin (one of the mediators of both erythrocyte and leukocyte adhesion in animal models of vaso-occlusion), was effective in reducing VOC frequency.48,49 However, it did not affect hematologic parameters or quality of life.
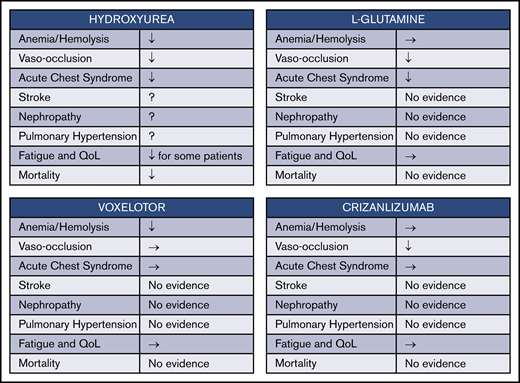
Effects of FDA-approved drugs for SCD. Four currently approved drugs for SCD each have distinct mechanisms and incompletely overlapping benefits. ↓, evidence for decrease in degree or frequency; →, evidence for no effect; QoL, quality of life; Fatigue and QoL ↓, evidence that fatigue is decreased and QoL is improved.
Effects of FDA-approved drugs for SCD. Four currently approved drugs for SCD each have distinct mechanisms and incompletely overlapping benefits. ↓, evidence for decrease in degree or frequency; →, evidence for no effect; QoL, quality of life; Fatigue and QoL ↓, evidence that fatigue is decreased and QoL is improved.
Furthermore, none of these 3 newer agents has been comprehensively studied in multiple age groups for an extended period of time. l-glutamine was studied in both children aged ≥5 years and adults, but the follow-up period was <1 year. Voxelotor was studied at 2 dose levels in patients aged >12 years for up to 72 weeks, although most end points were reported only after 24 weeks of treatment. Crizanlizumab was likewise administered over 52 weeks only to patients aged ≥16 years. In summary, therefore, all 3 drugs together have been studied in <500 persons receiving study drug (rather than placebo), and no long-term follow-up, particularly regarding end-organ damage and mortality, is available for any of them.
Thus, to achieve further therapeutic progress, we need to continue to develop a better understanding of mechanisms leading to organ dysfunction associated with poor quality of life and shortened survival, devise an evidence-based assessment system of risk for acute and chronic organ dysfunction, and adopt multi-agent therapy based on a better understanding of risk factors and disease mechanisms. Although vaso-occlusion occurs throughout the body, processes contributing to specific types of organ damage may vary. We need to understand the organ-specific characteristics of these events so that we can identify druggable targets and develop drugs and treatment modalities addressing those targets. With that point of view, and considering the pathophysiologic pathways shown in Figure 1, we might find additional drugs, either already FDA-approved drugs, such as anticoagulants, or drugs still in development, such as pyruvate kinase activators, that can be beneficial in combination with hydroxyurea, either with or without the other drugs shown in Figure 2. However, given the limitations of studies to date, insufficient data are likely to make difficult both the choices of multi-drug regimens as well as how to judge their efficacy and safety.
Treatment of acute complications of SCD to prevent morbidity and mortality
Acute events continue to bring high risk of morbidity and mortality in SCD. Among those seen with high frequency, acute chest syndrome remains the most common acute event leading to mortality in both children and adults.38,50
Acute chest syndrome typically presents with hypoxemia and pulmonary infiltrates consistent with pneumonia on chest radiography. This syndrome can have a rapid downhill course, which may result from the effect hypoxia has on the pulmonary endothelium, including upregulation of adhesion molecules and downregulation of protective mechanisms. Recently, in its approval of l-glutamine, the FDA cited not only its ability to reduce the frequency of VOC but also its efficacy in reducing the incidence of acute chest syndrome.45 Nonetheless, we cannot currently offer our patients pharmacologic therapies for acute chest syndrome. At this time, treatment usually consists of oxygen supplementation (and ventilatory support when needed), antibiotics (despite the paucity of evidence that acute chest syndrome often involves bacterial infection38 ), and blood transfusion. The latter, although never tested in a randomized clinical trial, seems to be the most effective treatment we have; simple transfusion is usually used in mild cases, and RBC exchange is used in severe cases, as well as in milder cases38 in patients with relatively higher baseline hemoglobin levels, such as with HbSC.
Over the past several years, investigators have developed a better understanding of acute chest syndrome that may lead to real therapeutic advances. Etiologic factors are not identifiable in nearly 50% of cases,38 although intravascular sickling,51 fat emboli, hypoxia,52 microvascular in situ thrombosis,52 infection,53 and acute painful VOC episodes may precede acute chest syndrome. Ghosh et al54 created an animal model of acute chest syndrome that relies on hemolysis as the precipitating factor. They note that available data support the idea that, whatever the cause, acute chest syndrome is most often preceded by an increase in hemolysis and thus in free heme.50,52,55 Research with this animal model has suggested both new pathophysiologic pathways as well as possible new treatment modalities. The authors found that pharmacologic inhibition of Toll-like receptor 4 (TLR4), as well as infusion of hemopexin before hemin infusion, protected sickle mice from developing acute chest syndrome.54
In somewhat similar studies, Belcher et al56 showed that infusion of hemoglobin or heme triggered vaso-occlusion in sickle mice, and that this effect was blocked by infusion of haptoglobin or the heme-binding protein hemopexin. They also found that blockade of TLR4 was protective, and that TLR4 knockout mice transplanted with sickle bone marrow were resistant to heme-induced vaso-occlusion. These groups have also shown that agents which interfere with cell adhesion or that increase antioxidant stress capacity also protect against the deleterious consequences of hemin infusion.56-58 These studies are examples of how modeling of specific complications of SCD may lead to promising therapeutic investigations. Indeed, l-glutamine, which is believed to improve antioxidant capacity, was approved by the FDA in part due to the reduction in acute chest syndrome seen with the drug.45,59 Animal models suggest that improvement in antioxidant response helps prevent heme oxygenase decline and thereby facilitates the breakdown of heme and reduction of its downstream toxic effects.58
Contribution of chronic end-organ damage to mortality
Clinically apparent chronic end-organ damage has a strong negative effect on overall survival in SCD, especially among adults. Several recent studies of survival in US SCD cohorts have documented that both the overall number of organs affected as well as specific types of end-organ damage strongly affect life expectancy. Chaturvedi et al60 showed that the presence of more than one organ affected by SCD greatly reduced survival from 14.0 to 7.8 years in adults, even after adjusting for age at enrollment, sex, sickle cell genotype (sickle cell anemia vs other), and hydroxyurea therapy. Elmariah et al39 showed that a history of central nervous system events, including stroke and seizure, as well as elevated tricuspid regurgitant jet velocity (emblematic of pulmonary hypertension), and sickle cell nephropathy have especially negative effects on survival; requirement for hospital admission for VOC in the past year, as well as several other clinical characteristics, are also associated with a higher risk of mortality. Four or more VOC episodes per year are especially strongly correlated to mortality.
Sickle cell nephropathy and pulmonary hypertension, at least as indicated by high tricuspid regurgitant jet velocity measured by echocardiogram, are 2 types of chronic end-organ damage that are highly linked to accelerated mortality. Nephropathy is highly associated with premature mortality (hazard ratio, 1.86), and survival with chronic kidney disease is severely limited.39 SCD affects both glomerular and tubular function of the kidney. Hyposthenuria, the inability to concentrate urine to >450 mOsm/kg with water deprivation, is seen in infancy and persists throughout life. Hyperfiltration, often defined as a measured glomerular filtration rate (GFR) >110 mL/min/1.73 m2, is present in >50% of infants from 9 months to 1 year of age42 and increases during childhood and into young adulthood. Nearly all adults age 18 to 30 years also exhibit high creatinine-based estimated GFR (130 mL/min/1.73 m2).61 Hyperfiltration also seems to result in glomerular injury, and hyperfiltration (estimated GFR >130-140 mL/min/1.73 m2) is associated with the development of microalbuminuria. Progression to macroalbuminuria affects nearly 40% of adults with HbSS by age 40 years.42 Tubular functional defects, in addition to hyposthenuria, have recently been shown to include impaired urinary acidification and distal tubular acidosis.62 Macroalbuminuria is a common finding (26%-28%) in SCD and is a known risk factor for decline in renal function. Current SCD guidelines suggest that any patient with macroalbuminuria or modest elevations in creatinine (>1.0 mg/dL in adults) should be considered as having significant renal disease and receive treatment with an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy.63 However, these therapies are used in analogy with other causes of proteinuria, although their true efficacy in SCD is uncertain. To study sickle cell nephropathy and potential therapeutic strategies, we first have to improve our understanding of risk factors and rate of progression. A growing body of evidence suggests that the rate of annual GFR decline is predictive of progression to end-stage renal disease. In a retrospective study, Xu et al64 asked: Can we identify SCD patients with a high risk of rapid GFR decline, to target therapies to this population and optimize the power of clinical studies to detect efficacy? Their study was able to identify 7 variables that increased the risk of rapid renal functional decline, including hemoglobin genotype (eg, HbSS/Sβ0 vs HbSC/Sβ+), presence of proteinuria, higher platelet count, higher reticulocyte count, higher systolic blood pressure, and lower body mass index. These and other factors, including the ApoL1 genetic variants thoroughly demonstrated to be risk factors for sickle nephropathy,65,66 must now be verified prospectively, so that future therapeutic trials can most efficiently identify valuable pharmacologic approaches by studying the proportion of patients at highest risk for significant renal disease.
Multi-agent therapy for SCD
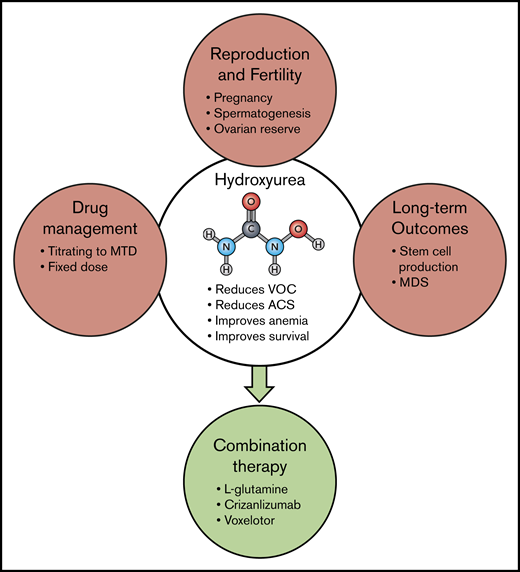
Although curative approaches such as gene therapy may eventually be a universally applicable way to cure SCD, the advances needed to make gene therapy simpler and transportable to resource-poor countries are likely many years in the future and possibly beyond the lifespans of the majority of those already living with the disease. Hydroxyurea, which inhibits HbS polymerization by inducing HbF expression and has multiple, potential other modes of action as well, has been very effective in altering the course of SCD for many patients63,67-71 ; however, its multiple effects also pose a spectrum of challenges for patients and caregivers (Figure 3). Despite higher HbF levels, even “responders” may still experience debilitating episodes of vaso-occlusion, strokes, organ dysfunction, and pain. Hydroxyurea is not recommended for use in female or male patients while they are planning to conceive children and may also reduce ovarian reserve. Hydroxyurea may also cause long-term damage to hematopoietic cells, reducing stem cell number and in some cases causing myelodysplasia.72 Physicians in most cases have managed hydroxyurea by titrating dosing to maximum tolerated dose, followed by frequent blood count checks to ensure safety. Only recently have studies shown that a standard dose can be both safe and beneficial.70,73-75
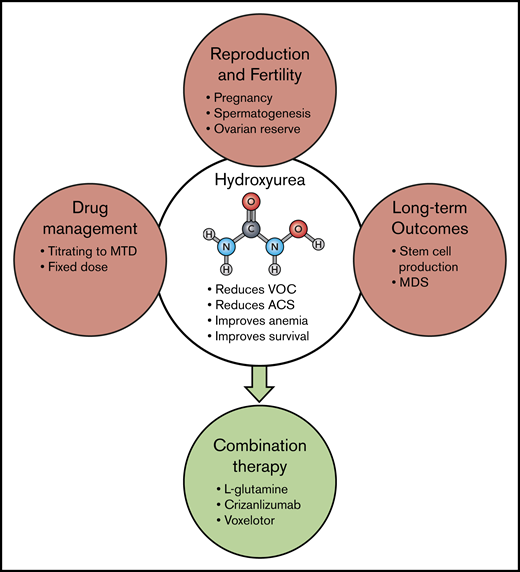
Hydroxyurea and multi-agent therapy. Although hydroxyurea (HU) has been used for decades in a variety of diseases, and its overall safety is well documented, there are still some unresolved questions regarding its long-term use. Guidelines continue to advise against HU when male or female patients plan to conceive a child, and questions remain about the effect of HU on spermatogenesis77,78 and ovarian reserve. In industrialized countries, management of HU treatment most often involves titration to maximum tolerated dose (MTD), but recent studies in Africa have documented the feasibility and benefit of fixed dosing without such resource-intensive monitoring.70,73,75 Concerns also persist regarding the possibility that long-term HU therapy reduces the number and viability of pluripotent stem cells and may predispose to myelodysplasia.72 The efficacy of HU in preventing organ damage, such as cardiac dysfunction, also remains uncertain. Now, to improve overall SCD therapy, we need to learn how to prescribe HU in combination with other, differently targeted therapies, such as those already FDA approved. Hopefully, additional therapies with additive or different therapeutic effects will be added to this list in the future. ACS, acute chest syndrome; MDS, myelodysplastic syndrome.
Hydroxyurea and multi-agent therapy. Although hydroxyurea (HU) has been used for decades in a variety of diseases, and its overall safety is well documented, there are still some unresolved questions regarding its long-term use. Guidelines continue to advise against HU when male or female patients plan to conceive a child, and questions remain about the effect of HU on spermatogenesis77,78 and ovarian reserve. In industrialized countries, management of HU treatment most often involves titration to maximum tolerated dose (MTD), but recent studies in Africa have documented the feasibility and benefit of fixed dosing without such resource-intensive monitoring.70,73,75 Concerns also persist regarding the possibility that long-term HU therapy reduces the number and viability of pluripotent stem cells and may predispose to myelodysplasia.72 The efficacy of HU in preventing organ damage, such as cardiac dysfunction, also remains uncertain. Now, to improve overall SCD therapy, we need to learn how to prescribe HU in combination with other, differently targeted therapies, such as those already FDA approved. Hopefully, additional therapies with additive or different therapeutic effects will be added to this list in the future. ACS, acute chest syndrome; MDS, myelodysplastic syndrome.
Three other drugs have recently been approved for SCD treatment, although each has only limited effects currently documented by data (Figure 2). Each of the newly approved drugs seems effective when used alone or concurrently with hydroxyurea. Moreover, each of these new drugs targets different pathophysiologic processes, and more differently targeted drugs are in the pipeline.
In this new world, however, we have yet to learn how to approach multi-agent therapy for SCD. The order of addition of each agent to treat a specific patient will need to be guided by response to previous therapy, risk factors identified for specific disease outcomes, and clinical studies to determine more thoroughly how the 4 currently approved drugs “mix and match.” This goal will have to be accomplished with an improved understanding of both disease pathophysiology and drug mechanisms of action, as well as defined end points in mind. Depending in part on the drugs being studied, appropriate end points may need to include HbF content; hemoglobin p50; markers of oxidative stress; inflammatory markers; white blood cell count, RBC, platelet, and reticulocyte counts; markers of hemolysis, including such measures as plasma free hemoglobin, and haptoglobin and hemopexin levels; frequency and duration of pain episodes; presence of end-organ damage and risk assessment; and quality-of-life measures. To date, however, although a variety of biomarkers predict higher mortality, only transcranial Doppler measurements have correlated with both prediction of risk as well as a therapeutic outcome (prevention of stroke).76
The health of millions of patients living with SCD today depends on near-term success in developing and delivering targeted and multi-agent therapeutic approaches to the sequelae of having RBCs with predominantly HbS. In the long run, and perhaps not during the lives of most patients with SCD alive today, gene therapy will become an easily accomplished and economically feasible cure. Although Judah Folkman, a true pioneer in medicine, cited E. L. Doctorow, who had originally referred to the process of writing, Folkman found it equally true of research. He said it is “like driving at night. You cannot see beyond the headlights, but you can make the whole trip that way.”79 We may not now be able to see exactly where we will end up, but we can nevertheless get there and save lives along the way.
Acknowledgments
M.J.T. acknowledges research support from the Doris Duke Charitable Foundation and from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (DK110104).
Authorship
Contribution: This manuscript was conceived and solely written by M.J.T.
Conflict-of-interest disclosure: M.J.T. has received research funds in support of basic research from Forma Therapeutics and CSL Behring; has conducted clinical research sponsored by Pfizer Inc. and Forma Therapeutics; and serves on the Executive Committee of the Cure Sickle Cell Initiative of the National Institutes of Health and on a Data Monitoring Committee for a study of crizanlizumab sponsored by Novartis.
Correspondence: Marilyn J. Telen, School of Medicine, Duke University, Box 2615, DUMC MSRB1, Research Dr, Durham, NC 27710; e-mail: marilyn.telen@duke.edu.