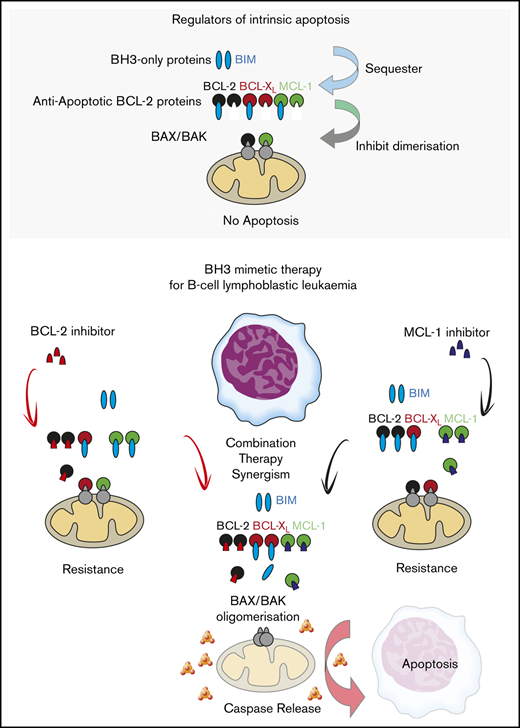
Key Points
BCL-2 and MCL-1 inhibitors synergize in vitro and demonstrate potent activity in vivo against high-risk B-ALL patient-derived xenografts.
The combination can induce tumor lysis syndrome in PDX models, indicating that appropriate precautions will be needed in clinical trials.

Abstract
Improving survival outcomes in adult B-cell acute lymphoblastic leukemia (B-ALL) remains a clinical challenge. Relapsed disease has a poor prognosis despite the use of tyrosine kinase inhibitors (TKIs) for Philadelphia chromosome positive (Ph+ ALL) cases and immunotherapeutic approaches, including blinatumomab and chimeric antigen receptor T cells. Targeting aberrant cell survival pathways with selective small molecule BH3-mimetic inhibitors of BCL-2 (venetoclax, S55746), BCL-XL (A1331852), or MCL1 (S63845) is an emerging therapeutic option. We report that combined targeting of BCL-2 and MCL1 is synergistic in B-ALL in vitro. The combination demonstrated greater efficacy than standard chemotherapeutics and TKIs in primary samples from adult B-ALL with Ph+ ALL, Ph-like ALL, and other B-ALL. Moreover, combined BCL-2 or MCL1 inhibition with dasatinib showed potent killing in primary Ph+ B-ALL cases, but the BH3-mimetic combination appeared superior in vitro in a variety of Ph-like ALL samples. In PDX models, combined BCL-2 and MCL1 targeting eradicated ALL from Ph− and Ph+ B-ALL cases, although fatal tumor lysis was observed in some instances of high tumor burden. We conclude that a dual BH3-mimetic approach is highly effective in diverse models of high-risk human B-ALL and warrants assessment in clinical trials that incorporate tumor lysis precautions.
Introduction
Although cure rates in pediatric precursor B acute lymphoblastic leukemia (B-ALL) approach 90%, only 30% to 40% of adult patients achieve long-term remission.1 Targeting antiapoptotic BCL-2 family proteins (BCL-2,2-4 BCL-XL,5 MCL-16 ) is an emerging therapeutic strategy in a range of leukemias. The selective BCL-2 inhibitor venetoclax has shown impressive efficacy in chronic lymphocytic leukemia3 and acute myeloid leukemia (AML),7 and in combination with BCL-XL inhibition (via navitoclax) in pediatric and adult relapsed or refractory B-ALL.8 S64315, a selective MCL-1 inhibitor and derivative of S63845,9 has entered clinical trials for AML, multiple myeloma, and lymphoma. BCL-2 and MCL-1 have been identified as important prosurvival proteins across multiple acute lymphoblastic leukemia (ALL) subtypes, including MLL-rearranged (KMT2Ar) ALL9-11 and BCR-ABL1 (Ph+) ALL,12 where combination ABL1 and BCL-2 inhibition appears synergistic in patient-derived xenograft (PDX) models.13,14 We have previously shown that S63845 is better tolerated by normal human CD34+ progenitor cells15,16 than standard cytotoxic drug combinations, and combined targeting of BCL-2 and MCL-1 was synergistic in preclinical AML models.16 Here, we have explored the efficacy of combined BCL-2 and MCL-1 inhibitors in preclinical models of BCR-ABL1 positive (Ph+), Ph-like (BCR-ABL1 negative ALL with kinase activating genetic alterations and blast gene expression profile similar to BCR-ABL1 ALL),17 and other subtypes of human B-ALL (Ph−).
Methods
In vitro cytotoxicity and drug synergy assays
Primary B-ALL patient BM samples (blasts >60%) and cell lines were incubated in drug concentrations between 1 nM and 10 μM for 48 hours or 96 hours, respectively. Cell viability was determined by SYTOX Blue Dead Cell Stain (S34857; Life Technologies) using an LSR-Fortessa (BD), and fluorescence-activated cell sorting data were analyzed using FlowJo as previously described.14,18
Event-free survival assays
NSG mice were injected IV with 1 × 106 ALL cells. In vivo treatment commenced initially upon 5% hCD45+ cells in peripheral blood and 1% after animal ethics committee review following acute tumor lysis syndrome (ATLS) was observed with combination therapy. A minimum of 6 mice received each treatment: vehicle, venetoclax (oral gavage; 25 mg, 50 mg, or 100 mg/kg per day, 5 days per week for 3 weeks), S63845 (3 mg/kg, 6 mg/kg, 12.5 mg/kg, or 25 mg/kg IV, twice weekly for 3 weeks), and ruxolitinib (90 mg/kg per day, 5 days per week for 3 weeks) alone and in combination. Human leukemia burden in blood was measured weekly by flow cytometry, and mice were examined daily and euthanized if found to be unwell (rough coat, failure to thrive, decreased activity, panting, circling, grinding teeth). Peripheral blood blast levels were compared with gauge efficacy and >25% hCD45+CD19+ defined as an event after animal ethics committee review. Histology was performed using institutional and independent external (Cerberus) review.
Results and discussion
Combined targeting of BCL-2 and MCL-1 synergistically induces apoptosis in vitro in high-risk B-ALL
We investigated whether selective BCL-2 (venetoclax) or MCL-1 (S63845) inhibitors enhanced the efficacy of tyrosine kinase inhibitors (TKIs), standard of care chemotherapeutic agents, or selective BH3-mimetics in 7 B-ALL cell lines. Venetoclax, but not S63845, strongly potentiated the activity of all TKIs tested in Ph+ALL, including TKIs not generally regarded as ABL1 inhibitors (Figure 1A). In contrast, TKI potentiation was not seen with either BH3-mimetic in CRLF2-rearranged Ph-like ALL cell lines, despite increased BIM expression following TKI exposure in both Ph+ and Ph-like ALL cell lines (supplemental Figure 1A). Furthermore, venetoclax and S63845 enhanced the efficacy of dexamethasone across Ph+, Ph− (including KMT2Ar-ALL) and Ph-like cell lines, and primary patient samples (Figure 1A; supplemental Figure 1B). The addition of venetoclax or S63845 markedly potentiated the modest killing observed with targeting BCL-2, MCL-1, or BCL-XL alone, across almost all cell lines (Figure 1A), and equimolar combinations of BCL-2 and MCL-1 inhibitors demonstrated potent killing (50% lethal concentration [LC50] <100 nM) in 6 of 7 B-ALL cell lines (Figure 1A). This included both Ph+ and Ph-like cell lines, where every pairwise BH3-mimetic combination tested demonstrated more consistent and potent synergy than combining each BH3-mimetic with either TKI or steroids (Figure 1A). This was recapitulated in vitro in a panel of Ph+ and Ph-like ALL PDXs (supplemental Figure 1C; supplemental Table 2). BH3-mimetic combinations demonstrated especially potent synergy (Bliss Sums >1000) in Ph-like ALL (supplemental Figure 1C-D). Similarly, in primary B-ALL patient samples, a minority was sensitive to selective targeting of BCL-2, MCL-1, or BCL-XL alone; however, simultaneous targeting of MCL-1 and either BCL-2 (with venetoclax or S55746) or BCL-XL demonstrated potent killing in 15 of 24 and 9 of 20 of samples, respectively. Notably, >50% of Ph+ and Ph− primary ALL samples were sensitive to combined BCL-2 and MCL-1 inhibition (Figure 1B; supplemental Table 1).
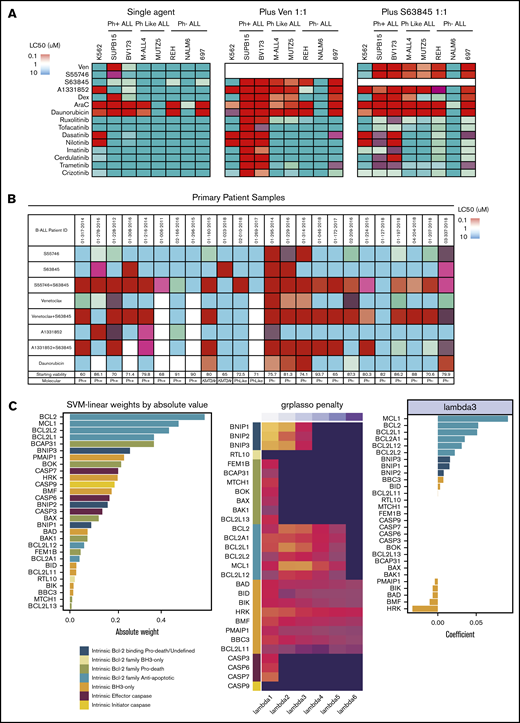
Interrogating prosurvival protein dependency in vitro in Ph+, Ph-like, and Ph− B-ALL using BH3-mimetic drugs alone and in combination. (A) Heat-map comparison of B-ALL cell-line sensitivity (LC50) to BH3-mimetics, chemotherapy agents, and tyrosine kinase inhibitors as a monotherapy, or in combination (tested in 1:1 ratio) after 48-hour exposure (see also supplemental Figure 1). (B) Heat-map comparison of primary B-ALL sensitivity (LC50) to BH3-mimetic monotherapy, or drug combinations (tested in 1:1 ratio), relative to chemotherapy (daunorubicin) after 48-hour exposure (n = 24). The control cell viability of each B-ALL sample after 48 hours in dimethyl sulfoxide is shown. For panels A and B, a color bar grading the LC50 values for each drug in the heat map is shown. (C) Transcriptional profiling and computational modeling of primary B-ALL patient samples. (left) Absolute weight of BCL-2 and BH3-only gene families in the trained Support Vector Classifier ordered by magnitude with the absolute value representing the importance of the gene to the resistant phenotype (BCL-2+MCL-1 inhibitor LC50 >100 nM). (middle) Heat map of coefficients estimated by the logistic regression model with group lasso penalty. Dark value is 0 coefficient (no importance) with brighter values representing a large coefficient and importance to the resistant phenotype by logistic regression. BCL-2 prosurvival and BH3-only prodeath genes demonstrated the most robust contribution to resistance in the model at the largest values of λ. Coefficients are transformed (absolute values raised to the power of 0.2) to enhance visualization. (right) Original coefficients (for λ = 3) in the linear regression model are given in the bar plot demonstrating directionality of gene expression to the resistant phenotype. The coefficients are positive for the BCL-2 prosurvival family genes, including BCL-2, MCL-1, BCL2L2 (BCL-W), and BCL2L1 (BCL-X), indicating a positive association between the level of expression of these genes with the resistant phenotype, whereas coefficients are negative for the BH3-only proapoptotic family genes HRK, BMF, BAD, BIK, PMAIP1, indicating a negative association between the level of expression of these BH3-only genes and resistance (see supplemental Figure 3). SVM, support vector machine.
Interrogating prosurvival protein dependency in vitro in Ph+, Ph-like, and Ph− B-ALL using BH3-mimetic drugs alone and in combination. (A) Heat-map comparison of B-ALL cell-line sensitivity (LC50) to BH3-mimetics, chemotherapy agents, and tyrosine kinase inhibitors as a monotherapy, or in combination (tested in 1:1 ratio) after 48-hour exposure (see also supplemental Figure 1). (B) Heat-map comparison of primary B-ALL sensitivity (LC50) to BH3-mimetic monotherapy, or drug combinations (tested in 1:1 ratio), relative to chemotherapy (daunorubicin) after 48-hour exposure (n = 24). The control cell viability of each B-ALL sample after 48 hours in dimethyl sulfoxide is shown. For panels A and B, a color bar grading the LC50 values for each drug in the heat map is shown. (C) Transcriptional profiling and computational modeling of primary B-ALL patient samples. (left) Absolute weight of BCL-2 and BH3-only gene families in the trained Support Vector Classifier ordered by magnitude with the absolute value representing the importance of the gene to the resistant phenotype (BCL-2+MCL-1 inhibitor LC50 >100 nM). (middle) Heat map of coefficients estimated by the logistic regression model with group lasso penalty. Dark value is 0 coefficient (no importance) with brighter values representing a large coefficient and importance to the resistant phenotype by logistic regression. BCL-2 prosurvival and BH3-only prodeath genes demonstrated the most robust contribution to resistance in the model at the largest values of λ. Coefficients are transformed (absolute values raised to the power of 0.2) to enhance visualization. (right) Original coefficients (for λ = 3) in the linear regression model are given in the bar plot demonstrating directionality of gene expression to the resistant phenotype. The coefficients are positive for the BCL-2 prosurvival family genes, including BCL-2, MCL-1, BCL2L2 (BCL-W), and BCL2L1 (BCL-X), indicating a positive association between the level of expression of these genes with the resistant phenotype, whereas coefficients are negative for the BH3-only proapoptotic family genes HRK, BMF, BAD, BIK, PMAIP1, indicating a negative association between the level of expression of these BH3-only genes and resistance (see supplemental Figure 3). SVM, support vector machine.
Transcriptional profiling of B-ALL samples and computational modeling identifies potential mediators of resistance to combined BCL-2 and MCL-1 inhibition
To understand whether the LC50 of primary ALL samples to combined BCL-2 and MCL-1 inhibition was predicted by the expression of BCL-2 family genes, samples were classified as sensitive (LC50 <10 nM) or resistant (LC50 >100 nM), and a Support Vector Classifier approach and logistic regression with group lasso penalty applied to log2 CPM RNA-seq values (Figure 1C; supplemental Figure 2). These 2 independent approaches identified that pretreatment expression levels of antiapoptotic BCL-2, MCL1, BCL2L1, and BCL2L12 correlated with resistance, while expression of proapoptotic BH3-only genes HRK, BMF, BAD, BIK, and PMAIP1 correlated with sensitivity.
Combined BCL-2 and MCL-1 inhibition in vivo has potent antileukemia activity in high-risk B-ALL PDXs
We next tested escalating in vivo doses of both venetoclax and S63845 in combination in nonleukemia-bearing NSG mice. Venetoclax up to 100 mg/kg with S63845 up to 12.5 mg/kg was well tolerated with no impact on blood counts or weight (supplemental Figure 3). However, in the cohorts receiving S63845 25 mg/kg with venetoclax at 3 dose levels (25/50/100 mg/kg per day), 7 of 12 mice in the lower-dose cohorts died with postmortem findings demonstrating bone marrow hypocellularity that was discordant with normal blood counts, and multiorgan edema, but precise causality could not be ascribed. All 6 mice receiving the highest doses (venetoclax 100 mg/kg and S63845 25 mg/kg) remained well. Although the latter suggests the deaths may not have been dose related, S63845 was limited to 12.5 mg/kg in combination therapy by the institutional animal ethics committee for the subsequent efficacy experiments.
To validate the observed efficacy of BCL-2 and MCL-1 inhibition in vivo, B-ALL PDX models were treated with a BCL-2 inhibitor (venetoclax or S55746), alone or combined with S63845, and/or kinase inhibitor if a targetable lesion was present. First, efficacy was evaluated in Ph+ and Ph− ALL PDXs after 5 days of treatment. Combined BCL-2 and MCL-1 inhibition demonstrated potent activity in the 2 Ph+ALL PDXs and was as potent as dasatinib alone or in combination with BCL-2 inhibition (Figure 2A; supplemental Figure 4A) while being well tolerated (supplemental Figure 4B-C).
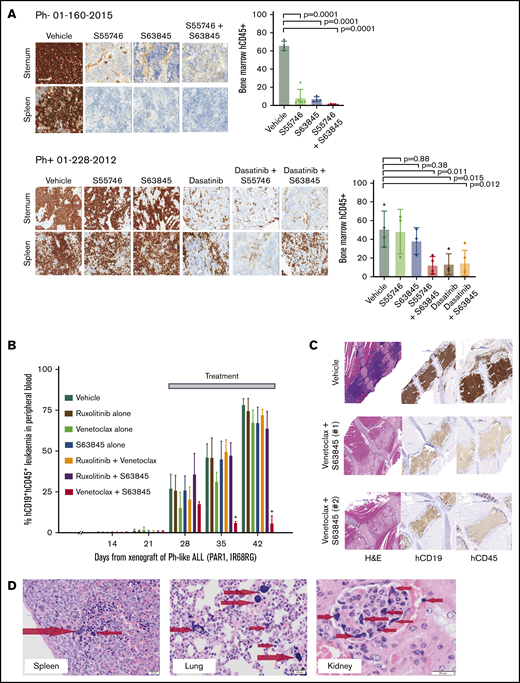
Interrogating prosurvival protein dependency in vivo in Ph+, Ph-like, and Ph− B-ALL PDX using BH3-mimetic drugs alone and in combination. (A) Irradiated NSG mice were transplanted with 1 × 106 primary B-ALL cells (Ph− 01-160-2015 or Ph+ 01-228-2012). Engraftment was confirmed at 6 weeks by detection of hCD45 in peripheral blood. Cohorts of 4 mice per group were then treated with (i) vehicle, (ii) S55746 (BCL-2 inhibitor), (iii) S63845 (MCL-1 inhibitor) or (iv) S55746 combined with S63845, and (v) dasatinib or (vi) dasatinib + S55746 (for 01-228-2012). Mice were euthanized on day 8, and immunohistological analysis for hCD45+ was performed on sternal bone marrow and spleen for infiltration by B-ALL cells captured at 100× magnification using the Aperio ScanScope. Representative cases from each cohort are shown. Flow cytometric analysis of flushed femurs showing the percentage of human CD45+ cells from B-ALL 01-160-2015 or 01-228-2012 after indicated treatments (mean ± 1 standard deviation and individual mouse values shown). B-ALL 01-160-2015 vehicle (n = 4), S63845 (n = 4), S55746 (n = 5), S55746+S63845 (n = 5). B-ALL 01-228-2012 vehicle (n = 4), S63845 (n = 3), S55746 (n = 3), S55746+S63845 (n = 4), dasatinib (n = 3), dasatinib + S63845 (n = 4). (B-C) NSG mice were transplanted with 1 × 106 Ph-like ALL cells (P2RY8-CRLF2 + JAK2 IR682RG). Leukemia engraftment and progression were assessed in groups of 6 female mice per treatment arm as shown with dosage regimen described in the “Methods.” (B) Treatment began when the mice engrafted 1% to 5% and human CD45+ cells and CD19+ proportion in peripheral blood (% hCD45+CD19+) were monitored weekly. *P = 0.0024 for Venetoclax + S63845 compared to vehicle by Mantel-Cox test. (C) Representative recipient sternal sections stained for hCD19 and hCD45 after treatment with vehicle (top) or Venetoclax + S63845 (middle and bottom, 2 independent recipients). Images taken at 20× magnification using the Panoramic Scan II by 3D Histech. (D) When venetoclax and S63845 were combined, fatal murine ATLS was observed with multiorgan involvement consisting of widely disseminated microemboli composed of nuclear and cytoplasmic debris derived from lysed leukemia cells, seen as deeply basophilic homogenous material as indicated by the arrows in spleen, lung, and kidney glomerulus.17,18 Images captured using the Olympus Model BX41 and Olympus U-CMAD 3 camera at 200× magnification (spleen and lung) and 400× magnification (kidney). H&E, hematoxylin and eosin.
Interrogating prosurvival protein dependency in vivo in Ph+, Ph-like, and Ph− B-ALL PDX using BH3-mimetic drugs alone and in combination. (A) Irradiated NSG mice were transplanted with 1 × 106 primary B-ALL cells (Ph− 01-160-2015 or Ph+ 01-228-2012). Engraftment was confirmed at 6 weeks by detection of hCD45 in peripheral blood. Cohorts of 4 mice per group were then treated with (i) vehicle, (ii) S55746 (BCL-2 inhibitor), (iii) S63845 (MCL-1 inhibitor) or (iv) S55746 combined with S63845, and (v) dasatinib or (vi) dasatinib + S55746 (for 01-228-2012). Mice were euthanized on day 8, and immunohistological analysis for hCD45+ was performed on sternal bone marrow and spleen for infiltration by B-ALL cells captured at 100× magnification using the Aperio ScanScope. Representative cases from each cohort are shown. Flow cytometric analysis of flushed femurs showing the percentage of human CD45+ cells from B-ALL 01-160-2015 or 01-228-2012 after indicated treatments (mean ± 1 standard deviation and individual mouse values shown). B-ALL 01-160-2015 vehicle (n = 4), S63845 (n = 4), S55746 (n = 5), S55746+S63845 (n = 5). B-ALL 01-228-2012 vehicle (n = 4), S63845 (n = 3), S55746 (n = 3), S55746+S63845 (n = 4), dasatinib (n = 3), dasatinib + S63845 (n = 4). (B-C) NSG mice were transplanted with 1 × 106 Ph-like ALL cells (P2RY8-CRLF2 + JAK2 IR682RG). Leukemia engraftment and progression were assessed in groups of 6 female mice per treatment arm as shown with dosage regimen described in the “Methods.” (B) Treatment began when the mice engrafted 1% to 5% and human CD45+ cells and CD19+ proportion in peripheral blood (% hCD45+CD19+) were monitored weekly. *P = 0.0024 for Venetoclax + S63845 compared to vehicle by Mantel-Cox test. (C) Representative recipient sternal sections stained for hCD19 and hCD45 after treatment with vehicle (top) or Venetoclax + S63845 (middle and bottom, 2 independent recipients). Images taken at 20× magnification using the Panoramic Scan II by 3D Histech. (D) When venetoclax and S63845 were combined, fatal murine ATLS was observed with multiorgan involvement consisting of widely disseminated microemboli composed of nuclear and cytoplasmic debris derived from lysed leukemia cells, seen as deeply basophilic homogenous material as indicated by the arrows in spleen, lung, and kidney glomerulus.17,18 Images captured using the Olympus Model BX41 and Olympus U-CMAD 3 camera at 200× magnification (spleen and lung) and 400× magnification (kidney). H&E, hematoxylin and eosin.
We then treated cohorts of a Ph-like ALL PDX with P2RY8-CRLF2 and JAK2 mutation with different combinations of venetoclax, S63845, and ruxolitinib. Combined venetoclax and S63845 showed greater efficacy than each drug alone and other combinations (Figure 2B-C). Importantly, in 2 independent experiments, 4 of 21 mice had confirmed histological features of ATLS,19,20 having become unwell or dying within hours of receiving S63845 in addition to venetoclax (Figure 2D) with a high circulating tumor burden at time of treatment (supplemental Figure 5A).
To minimize the risk of ATLS in subsequent experiments, treatment was commenced at a lower tumor burden (median 1% engraftment) using lead-in and lower final doses of venetoclax. Using this regimen, venetoclax and S63845 combined was superior to either alone with statistically significant lower leukemia burden in the blood at assayed time points (supplemental Figure 5B) and longer event-free survival with leukemia burden <25% (supplemental Figure 5C).
In summary, we have demonstrated that dual BCL-2 and MCL-1 inhibition induces synergistic killing in vitro and rapid cytoreduction in vivo in several high-risk B-ALL models, including BCR-ABL1 and CRLF2-rearranged subtypes. Concurrent BCL-2 and MCL-1 inhibition was comparable or superior to dasatinib combined with each BH3-mimetic in Ph+ ALL and was highly active in subtypes of B-ALL for which no targetable kinase lesion was identified, with ATLS observed in some mice. Notably, concurrent BCL-2 and MCL-1 inhibition was superior to each BH3-mimetic combined with ruxolitinib in Ph-like B-ALL, where the latter agent demonstrated limited efficacy. Simultaneous BCL-2 and MCL-1 inhibition appears to have a novel breadth of potency across high-risk B-ALL subtypes, including Ph-like ALL, with resistance predicted by the expression level of BCL2 family antiapoptotic genes BCL-2, MCL-1, and BCL2L1. We conclude that this approach warrants clinical trials, which should include mandatory tumor lysis precautions and monitoring for hematologic and nonhematologic toxicity with combination therapy.
Send data sharing requests to Donia M. Moujalled (donia.moujalled@monash.edu) or Diane T. Hanna (hanna.d@wehi.edu.au).
Acknowledgments
This work was supported by the National Health and Medical Research Council program grant GNT1113577 (A.W.R.), project grants GNT1060179 and GNT1122783 (A.P.N.), Cancer Council Victoria, Victorian Cancer Agency, Leukaemia Foundation of Australia, Leukemia and Lymphoma Society (Specialized Center of Research) grant 20454683, Australian Cancer Research Foundation, Monash Partners, the Alfred Foundation, and the Medical Research Future Fund, Children's Cancer Foundation, The Department of Health and Human Services through the Victorian Cancer Agency (S.L.K.), as well as by philanthropic support from the Walter and Eliza Hall Institute, and operational infrastructure grants through the Australian Government Independent Research Institute Infrastructure Support Scheme and the Victorian Government Operational Infrastructure Support. This work has received funding support from Servier.
Authorship
Contribution: S.L.K., A.P.N., A.W., P.G.E., A.W.R., D.C.S.H., D.M.M., and D.T.H. conceptualized the study; S.L.K., A.P.N., A.W., P.G.E., D.M.M., D.T.H., S.H.-z., M.J.D., O.G., and L.R. provided the methodology; S.H.-z. and M.J.D. provided the software; S.L.K., A.P.N., A.W., P.G.E., A.W.R., D.C.S.H., D.M.M., and D.T.H. provided the validation; S.L.K., A.P.N., A.W., P.G.E., A.W.R., D.C.S.H., D.M.M., D.T.H., S.H.-z., and M.J.D. performed the formal analysis; D.M.M., D.T.H., A.G., L.B., C.A.W., S.H.-z., M.J.D., G.P., L.K.-B., R.B., and P.G.E. performed the investigation; C.G.M., M.C., S.B., V.L., S.F., A.-L.M., L.K.-B., M.S., G.L., O.G., L.R., and C.A.W. were responsible for the resources; D.M.M., D.T.H., S.L.K, A.P.N., A.W., A.W.R., C.G.M., S.H., and M.J.D. wrote the manuscript; D.M.M., D.T.H., S.H., S.L.K., and A.P.N. were responsible for the visualization; S.L.K., A.P.N., A.W., P.G.E., A.W.R., C.A.W., and D.C.S.H. supervised the study; S.L.K., A.P.N., and A.W. were responsible for project administration; and S.L.K., A.W., P.G.E., A.W.R., and D.C.S.H. acquired the funding.
Conflict-of-interest disclosure: A.G., A.P.N., A.W.R., C.A.W., D.C.S.H., G.L., S.H.-z., and M.J.D. are employees of the Walter and Eliza Hall Institute, which receives milestone and royalty payments related to venetoclax. A.W., D.M.M., and P.G.E. receive royalty payments related to venetoclax. A.W., A.W.R., D.M.M., and G.L. have received research funding from Servier. A.P.N., A.W.R., D.C.S.H., G.L., S.H.-z., S.L.K., and M.J.D. are recipients of a share in royalty payments paid to the Walter and Eliza Hall Institute of Medical Research. A.W.R. has received research funding from AbbVie, Genentech, Janssen, and BeiGene. C.G.M. has received research funding from AbbVie, Pfizer, Loxo Oncology, Amgen, and Illumina. G.L. has received research funding from Genentech and Anaxis Pharma. S.L.K. has received research funding from AbbVie, Novartis, Jazz Pharmaceuticals, and Bristol-Myers Squibb. A.-L.M., M.S., O.G., and S.B. are employees of Servier. The remaining authors declare no competing financial interests.
Correspondence: Donia M. Moujalled, Australian Centre for Blood Diseases, Australian Centre for Blood Diseases, Monash University, Level 1 Walkway via The Alfred Centre, 99 Commercial Rd, Melbourne, VIC 3004, Australia; e-mail: donia.moujalled@monash.edu.