Key Points
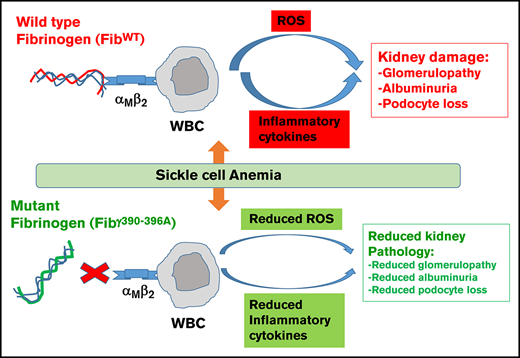
Elimination of the fibrinogen integrin αMβ2-binding motif improves renal pathology in SCA.
The fibrinogen γ390-396 motif represents a novel therapeutic target that may improve sickle cell disease outcomes.

Abstract
Sickle cell anemia (SCA) is caused by a point mutation in the β-globin gene that leads to devastating downstream consequences including chronic hemolytic anemia, episodic vascular occlusion, and cumulative organ damage resulting in death. SCA patients show coagulation activation and inflammation even in the absence of vascular occlusion. The coagulation factor fibrinogen is not only central to hemostasis but also plays important roles in pathologic inflammatory processes, in part by engaging neutrophils/macrophages through the αMβ2 integrin receptor. To determine whether fibrin(ogen)-mediated inflammation is a driver of SCA-associated pathologies, hematopoietic stem cells from Berkeley sickle mice were transplanted into homozygous Fibγ390-396A mice that express normal levels of a mutant form of fibrin(ogen) that does not engage αMβ2. Fibγ390-396A mice with SCA displayed an impressive reduction of reactive oxygen species (ROS) in white blood cells (WBCs), decreased circulating inflammatory cytokines/chemokines, and significantly improved SCA-associated glomerular pathology highlighted by reduced glomerulosclerosis, inflammatory cell infiltration, ischemic lesions, mesangial thickening, mesangial hypercellularity, and glomerular enlargement. In addition, Fibγ390-396A mice with SCA had improved glomerular protective responses and podocyte/mesangial transcriptional signatures that resulted in reduced albuminuria. Interestingly, the fibrinogen γ390-396A mutation had a negligible effect on cardiac, lung, and liver functions and pathologies in the context of SCA over a year-long observation period. Taken together, our data support that fibrinogen significantly contributes to WBC-driven inflammation and ROS production, which is a key driver of SCA-associated glomerulopathy, and may represent a novel therapeutic target against irreversible kidney damage in SCA.
Introduction
Sickle cell anemia (SCA) is a monogenic red blood cell (RBC) disorder affecting over 100 000 Americans and millions worldwide.1-4 SCA is caused by a point mutation in the β-globin gene, resulting in sickle hemoglobin (HbS). Repeated cycles of HbS sickling cause chronic hemolytic anemia, recurrent vaso-occlusion associated with severe pain and inflammation, and acute and cumulative organ damage that manifests as acute chest syndrome, pulmonary hypertension, stroke, sickle lung disease, nephropathy, end-stage renal disease, and eventual death due to cumulative organ failure.3,5-8 The collective health care costs of SCA are over 1 billion dollars per year in the United States alone.9
Improved prevention of infection-related deaths in early childhood due to newborn screening and early diagnosis, infection prophylaxis strategies, and overall better medical care has allowed SCA patients to survive well into adulthood.10 Hence, end-organ pathologies are now emerging as the most common cause of death. As a result, the lifespan of SCA patients has not changed for the last 3 decades, as deaths from end-organ failure are the most common cause of mortality. Beyond providing symptom relief and preventing infections, therapeutic options are limited to chronic transfusions, hydroxyurea (a drug that induces the fetal hemoglobin that prevents RBC sickling), or an allogenic bone marrow transplant (BMT), a curative therapy available only to 10% to 15% patients in the developed world.2,11 Hence, targeted therapies that prevent end-organ damage are much needed.
Vascular occlusion and endothelial dysfunction coupled with coagulation activation and inflammatory processes have been frequently proposed as significant drivers of SCA-associated pathologies.12,13 Platelet activation is commonly observed in SCA, and is shown to promote adhesion of sickle RBCs to vascular endothelium and may contribute to thrombosis and pulmonary hypertension in SCA.14,15 SCA patients show evidence of chronic coagulation activation even in the absence of vascular occlusion.16 The key coagulation factor thrombin bridges the hemostatic and inflammatory systems. Our group has recently shown that reducing prothrombin by pharmacologic and genetic approaches reduces kidney, heart, lung, and liver pathologies, and significantly prolongs the lifespan of sickle mice.17 One mechanism linking thrombin to SCA inflammation appears to be activation of protease-activated receptors18-20 to stimulate platelets,18,19 fibroblasts,21 and endothelial cells,22 which affect leukocyte trafficking and activation.23-25 However, analyses of fibrinogen as a direct downstream target of thrombin that mediates SCA disease pathologies has remained untested.
The coagulation factor fibrinogen is not only central to hemostasis but also plays important roles in pathologic inflammatory processes. Leukocytes can engage fibrinogen through the integrin receptor αMβ2 to stimulate leukocyte effector functions such as phagocytosis, degranulation, and the production of proinflammatory cytokines/chemokines, such as interleukin 6 (IL-6), tumor necrosis factor α (TNF-α), and IL-1.26-28 Fibγ390-396A mice express a mutant form of fibrinogen that has normal clotting function but does not support αMβ2-mediated cell adhesion, thereby reducing the fibrinogen-mediated inflammatory response.29 Indeed, Fibγ390-396A mice develop markedly reduced inflammatory disease in a number of contexts, including arthritis, colitis, and neuromuscular disease.29-32 To determine whether the fibrin(ogen)-inflammation axis promotes end-organ damage in SCA, we transplanted bone marrow hematopoietic stem cells (HSCs) from Berkeley sickle mice into wild-type (WT) fibrinogen and Fibγ390-396A mice. Analysis of WT and Fibγ390-396A mice with and without SCA over the course of 1 year revealed that the fibrinogen γ390-396 motif significantly contributes to sickle renal pathology while minimally affecting the development of pathologies in other organs.
Methods
Experimental mice
Berkeley sickle mice [Tg(Hu-miniLCRα1GγAγδβS)Hba0//Hba0Hbb0//Hbb0] that exclusively express human HbS33 and C57BL/6-inbred carrying selected knock-in mutations in the fibrinogen γ-chain (amino acids 390 to 396 were mutated to alanine), named Fibγ390-396A mice, were described.29 Mice expressing normal human hemoglobin (HbA) were generously provided by Cheryl Hillery (Medical College of Wisconsin, Milwaukee, WI). All experiments were conducted at Cincinnati Children’s Research Foundation Vivarium with appropriate institutional animal care and use committee approval (#2016-0041). Bone marrow cells from Berkeley mice expressing human HbS or healthy hemoglobin (HbA), or C57BL/6-Ly5.1 mice expressing mouse hemoglobin, were transplanted into WT fibrinogen (FibWT) or Fibγ390-396A recipients to generate chimeras: WT fibrinogen HbS (FibWT sickle chimera [SS]), Fibγ390-396A HbS (Fibγ390-396A SS), WT fibrinogen HbA (FibWT AA), Fibγ390-396A HbA (Fibγ390-396A AA), WT fibrinogen C57BL/6-Ly5.1 (FibWT B6), or Fibγ390-396A C57BL/6-Ly5.1 (Fibγ390-396A B6) (supplemental Methods; supplemental Figure 1).
Analyses of urine albumin and osmolality
Twenty-four-hour urine was collected from experimental mice using metabolic cages. A mouse albumin ELISA kit was used for determining the concentration of urine albumin (Bethyl Laboratories. Montgomery, TX) and urine creatinine concentration was measured with the alkaline picrate reagent kit (R&D Systems, Minneapolis, MN). Urine albumin concentration was normalized to creatinine concentration. Urine osmolality was measured using a Vapro Pressure Osmometer 5600 (Wescor Biomedical Systems, Logan, UT).
Histopathology analyses of organ samples
All mice were weighed before being euthanized. Kidney, heart, lung, and liver were fixed in 10% buffered formalin and embedded in paraffin. Sections (4 µm) were stained with hematoxylin and eosin (H&E), periodic acid–Schiff (PAS), and Masson's trichrome staining and evaluated by a pathologist blinded to animal genotype.
Statistical analyses
Data were analyzed using GraphPad Prism software version 8. Nonparametric Mann-Whitney U tests were used for analyses of statistical significance, unless otherwise stated. We have also used 1-way analysis of variance (ANOVA) followed by the Tukey's multiple comparison test (parametric) for 4 groups of data, the Dunnett's test (parametric) comparing all groups to the FibWT SS group, or the Dunn's test for nonparametric data where applicable. Values were expressed as mean ± standard error of mean (SEM).
Results
SCA in FibWT and Fibγ390-396A mice
Circulating fibrinogen is synthesized and originates exclusively from the liver. HSC transplant using bone marrow from mice carrying the SCA β-globin gene (ie, HbS) bypasses laborious and time-consuming breeding of sickle mice with WT or mutant fibrinogen mice. High-performance liquid chromatography–based hemoglobin analysis confirmed the HbS or HbA profile (supplemental Figure 2A-G) 4 months after HSC (HbS or HbA) transplant into FibWT or Fibγ390-396A mice. Only mice fully chimeric for donor HbS (SS) or HbA (AA) with a negligible level of endogenous mouse hemoglobin were considered evaluable and analyzed further. HSCs from C57BL/6-Ly5.1 (B6) mice were also transplanted into FibWT or Fibγ390-396A mice as transplant controls.
Complete blood count analyses of fully chimeric mice were done at 12 months post-BMT. No significant differences in RBC parameters were observed between FibWT SS and Fibγ390-396A SS mice, both showing the classic SCA RBC parameters; likewise, there were no differences between the RBC parameters of FibWT AA/B6 and Fibγ390-396A AA/B6 mice (supplemental Table 1), both groups showing normal RBC indices. As expected, significant differences were observed in RBC parameters between SS vs nonsickle chimeras (AA/B6). Consistent with development of a thromboinflammatory disease in SS mice, significantly increased white blood cell (WBC), neutrophil, and lymphocyte counts were observed in FibWT SS mice compared with FibWT AA/B6 or Fibγ390-396A AA/B6 mice. We also found significantly increased neutrophil and lymphocyte counts in FibWT SS mice compared with Fibγ390-396A AA/B6 mice. Neutrophil counts were significantly reduced in Fibγ390-396A SS mice compared with FibWT SS mice. A trend toward lower WBC, monocyte, and platelet counts was observed in Fibγ390-396A SS mice compared with FibWT SS mice, and Fibγ390-396A AA/B6 mice compared with FibWT AA/B6 mice (supplemental Table 1). However, we found lymphocyte counts were not affected by fibγ390-396A mutation. Overall, these data demonstrate that imposition of Fibγ390-396A mutation lowered WBC, neutrophil, monocyte, and platelet counts, suggesting reduced inflammation, but the fibrinogen mutation does not affect RBC parameters.
Fibγ390-396A SS mice display reduced ROS production in WBCs
Overall, enhanced reactive oxygen species (ROS) production was observed in circulating cells from SS compared with nonsickle chimeras, which is in line with published observations.34-36 SCA patients show higher oxidative stress than the healthy subjects.37 Although SCA is a RBC disorder, platelets and WBCs are involved in downstream disease pathogenesis.38,39 Accordingly, besides RBCs, we investigated the status of ROS production in WBCs and platelets in our experimental cohorts at 9 months posttransplant. Consistent with the idea that the fibrinogen γ390-396 motif can engage subsets of WBCs (eg, neutrophils and macrophages), we observed a significant decrease in ROS production in WBCs of Fibγ390-396A SS compared with FibWT SS mice (Figure 1B). In contrast, we observed a trend for reduced ROS production in RBCs (Figure 1A) and ROS production of platelets (Figure 1C) was not significantly reduced in Fibγ390-396A SS compared with FibWT SS mice.
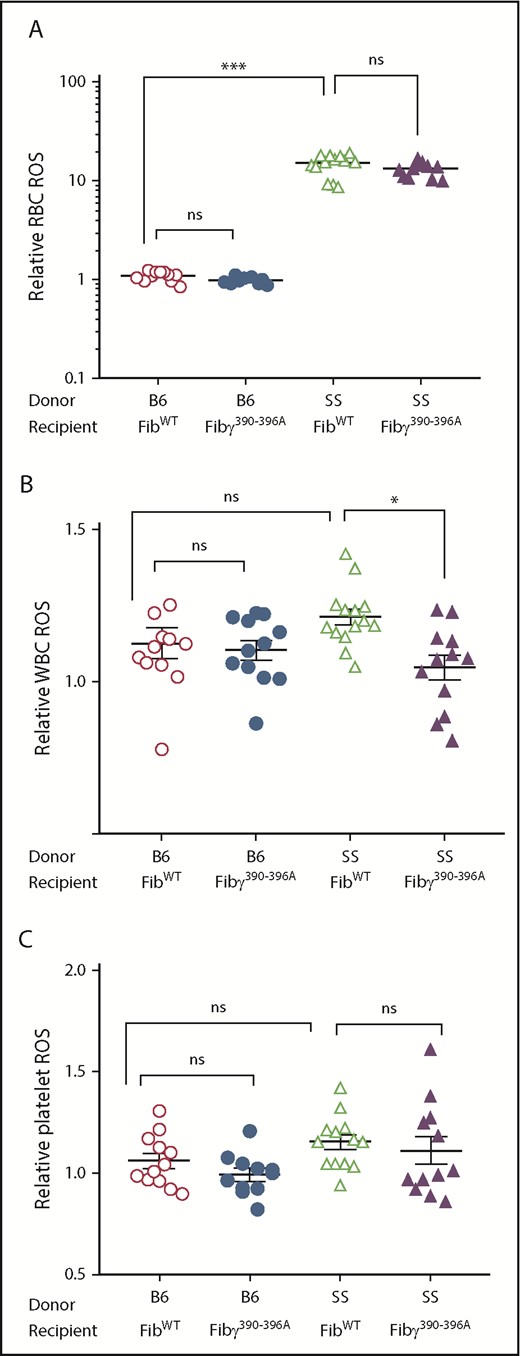
Relative ROS production of WBCs is reduced in Fibγ390-396ASS mice compared with the FibWTSS mice. Blood samples from the experimental mice were collected 9 months after BMT and stained with RBCs, WBCs, and platelet-specific antibodies and ROS detection reagent (CM-H2-DCFDA). (A) Relative RBC ROS. (B) Relative WBC ROS. (C) Relative platelets ROS. FibWT B6 (n = 12-14), Fibγ390-396A B6 (n = 11-14), FibWT SS (n = 12-14), and Fibγ390-396A SS (n = 12-14). Similar number of fully chimeric male and female mice were used in the experimental groups. Each symbol in the graph represents an individual mouse. The bars in the graph indicate the mean ± SEM. Statistical significance was determined using 1-way ANOVA followed by the Tukey's multiple comparison test; statistical significance is indicated as **P ≤ .01, ****P ≤ .0001. (A) For RBC ROS, the Dunn's test was used; ***P ≤ .001. ns, not significant.
Relative ROS production of WBCs is reduced in Fibγ390-396ASS mice compared with the FibWTSS mice. Blood samples from the experimental mice were collected 9 months after BMT and stained with RBCs, WBCs, and platelet-specific antibodies and ROS detection reagent (CM-H2-DCFDA). (A) Relative RBC ROS. (B) Relative WBC ROS. (C) Relative platelets ROS. FibWT B6 (n = 12-14), Fibγ390-396A B6 (n = 11-14), FibWT SS (n = 12-14), and Fibγ390-396A SS (n = 12-14). Similar number of fully chimeric male and female mice were used in the experimental groups. Each symbol in the graph represents an individual mouse. The bars in the graph indicate the mean ± SEM. Statistical significance was determined using 1-way ANOVA followed by the Tukey's multiple comparison test; statistical significance is indicated as **P ≤ .01, ****P ≤ .0001. (A) For RBC ROS, the Dunn's test was used; ***P ≤ .001. ns, not significant.
Reduction of inflammatory cytokines/chemokines in Fibγ390-396A SS mice
A host of studies have implicated a pathologic role of numerous cytokines/chemokines during steady-state and vaso-occlusive crisis in SCA patients.4,40 Mice with SCA show elevated plasma levels of numerous proinflammatory cytokines/chemokines.40 Accordingly, we noted elevated levels of IL-6, TNFα, and KC (CXCL-1, a functional homolog of human IL-8) in FibWT SS mice compared with FibWT AA mice (Figure 2). However, IL-6 was significantly reduced in Fibγ390-396A SS mice compared with the FibWT SS mice (Figure 2A). We also observed significantly reduced levels of TNF-α (Figure 2B), a trend toward reduction of KC (Figure 2C), IL-1α, and IL-10 (supplemental Figure 3A-B) in Fibγ390-396A SS mice compared with FibWT SS mice. However, we did not observe any significant reduction in macrophage inflammatory protein 1α, macrophage inflammatory protein 1β, IL-13, IL-5, IL-2, or soluble vascular cell adhesion molecule-1 concentration between Fibγ390-396A SS and FibWT SS mice (data not shown). Collectively, these data demonstrate that Fibγ390-396A SS mice have significantly reduced key circulating markers of inflammation compared with the FibWT SS mice.

Proinflammatory cytokines/chemokines are reduced in Fibγ390-396ASS mice compared with the FibWTSS mice. One year post-BMT, blood samples were collected from the experimental mice. Platelet-poor plasma samples were used for cytokine profiling by Luminex assay. (A) IL-6. (B) TNF-α. (C) KC. Similar number of fully chimeric male and female mice were used in the experimental groups. Each symbol in the graph represents an individual mouse. FibWT AA/B6 (n = 11-15), Fibγ390-396A AA/B6 (n = 8-12), FibWT SS (n = 17-22), and Fibγ390-396A SS (n = 17-22). The bars in the graph indicate the mean ± SEM. Statistical significance was determined using 1-way ANOVA followed by the Tukey's multiple comparison test; statistical significance is indicated as **P ≤ .01, *P ≤ .05.
Proinflammatory cytokines/chemokines are reduced in Fibγ390-396ASS mice compared with the FibWTSS mice. One year post-BMT, blood samples were collected from the experimental mice. Platelet-poor plasma samples were used for cytokine profiling by Luminex assay. (A) IL-6. (B) TNF-α. (C) KC. Similar number of fully chimeric male and female mice were used in the experimental groups. Each symbol in the graph represents an individual mouse. FibWT AA/B6 (n = 11-15), Fibγ390-396A AA/B6 (n = 8-12), FibWT SS (n = 17-22), and Fibγ390-396A SS (n = 17-22). The bars in the graph indicate the mean ± SEM. Statistical significance was determined using 1-way ANOVA followed by the Tukey's multiple comparison test; statistical significance is indicated as **P ≤ .01, *P ≤ .05.
Fibγ390-396A SS mice develop cardiac, lung, and liver pathologies similar to FibWT SS mice
Coagulation activation, endothelial dysfunction, and inflammation have been linked to pulmonary hypertension and ventricular diastolic dysfunction in SCA.12,17 To assess the effects of Fibγ390-396A mutation on cardiac function, echocardiography was performed 12 months post-BMT. As expected, FibWT SS mice demonstrated increased left ventricular (LV) diastolic internal dimension (LVID;d) due to anemia-related high-output physiology; a similar change was observed in Fibγ390-396A SS mice compared with FibWT AA/B6 mice (Figure 3A). Compared with FibWT AA/B6, FibWT SS mice also had a larger LV systolic internal dimension (LVID;s) (Figure 3B) and, accordingly, a lower LV fractional shortening (FS) (Figure 3C). We found significant reduction of LV hypertrophy (LVH) in Fibγ390-396A SS mice compared with FibWT SS, with the latter having the greatest LV mass among the 4 cohorts (Figure 3D). Table 1 shows that left atrial (LA) dimension was significantly higher in FibWT SS compared with both FibWT AA/B6 and Fibγ390-396A AA/B6 mice. The LA dimension was intermediate in Fibγ390-396A SS mice: slightly lowered compared with FibWT SS and slightly elevated compared with FibWT AA/B6 and Fibγ390-396A AA/B6 mice, but the differences were not significant. Ventricular parameters of diastolic interventricular septal thickness (IVS;d) and LV posterior wall thickness (LVPW;d) did not differ among 4 groups. There were no differences in diastolic parameters of mitral valve (MV) inflow velocity and tissue Doppler across the cohorts. Cardiac histopathology analyses revealed similar levels of fibrosis in both FibWT SS and Fibγ390-396A SS mice (supplemental Figure 4). Taken together, we found expected changes in LV dimensions due to sickle anemia in both of the SS groups. The FibWT SS mice had both LVH and LA dilation, which may be attributable to worse renal pathology in FibWT SS mice compared with Fibγ390-396A SS mice.
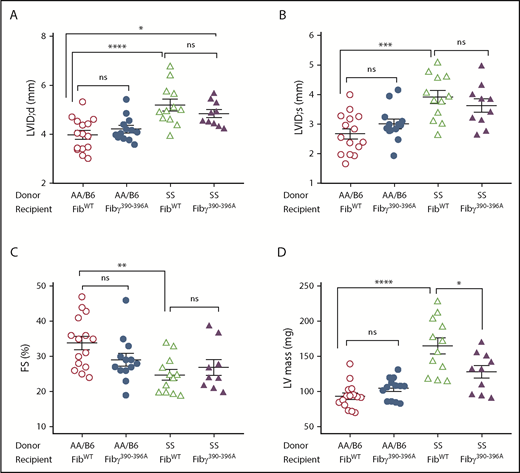
Comparison of LV dimensions, systolic function, and mass. (A-B) LVID;d and LVID;s, respectively. (C) LV FS. (D) Echocardiographically derived LV mass is reduced in Fibγ390-396A SS compared with the FibWT SS mice. Echocardiograms were performed on mixed-sex chimeric mice 12 months after BMT. Data are presented as mean ± SEM. Statistical significance was determined using 1-way ANOVA followed by the Tukey's multiple comparison test; statistical significance is indicated as *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Comparison of LV dimensions, systolic function, and mass. (A-B) LVID;d and LVID;s, respectively. (C) LV FS. (D) Echocardiographically derived LV mass is reduced in Fibγ390-396A SS compared with the FibWT SS mice. Echocardiograms were performed on mixed-sex chimeric mice 12 months after BMT. Data are presented as mean ± SEM. Statistical significance was determined using 1-way ANOVA followed by the Tukey's multiple comparison test; statistical significance is indicated as *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Pulmonary histology of our FibWT SS mice revealed the presence of occasional foamy alveolar macrophages, thickened arterial/arteriolar tunica media, increased lymphocyte and macrophage infiltration as well as edema around the blood vessel and vascular congestion (data not shown).17 However, no significant differences of these pathologies were observed in Fibγ390-396A SS mice (data not shown). As expected, FibWT AA/B6 and Fibγ390-396A AA/B6 chimeras were free of lung histopathological features (data not shown).
SCA hepatic pathologies occur due to inflammation, congestion, and coagulative necrosis.17 As expected, no histopathological events were observed in the liver of FibWT AA/B6 or Fibγ390-396A AA/B6 chimeras (data not shown). However, both FibWT SS and Fibγ390-396A SS mice showed a similar degree of multifocal coagulative hepatic necrosis, lymphocytic and macrophage infiltration in the peripheral hepatocytes, marked congestion within hepatic sinusoids with shrinkage of hepatocytes, and vasculature obstruction with sickle RBCs (data not shown).
Fibγ390-396A SS mice develop reduced sickle renal pathology
Nephropathy in SCA patients is a major complication that manifests as albuminuria in >50% of the patients and progresses to renal failure.7,41 Sickle mice show many histologic features similar to SCA patients.42 The kidney–to–body-weight ratio of our experimental mice did not show any significant differences between the FibWT AA/B6 vs Fibγ390-396A AA/B6 and FibWT SS vs Fibγ390-396A SS groups, as well as nonsickle vs sickle groups (supplemental Figure 5). Importantly, glomerular histopathology revealed that FibWT SS mice glomeruli exhibited glomerulosclerosis, inflammatory cell infiltration, ischemic lesions, mesangial thickening, mesangial hypercellularity, and glomerular enlargement (Figure 4 left panels); however, the glomeruli of Fibγ390-396A SS mice were notable for significantly reduced glomerular pathology (Figure 4 middle panels). Semiquantitative histopathological scoring of stained kidney sections confirmed a significant reduction in glomerular pathology in Fibγ390-396A SS mice compared with FibWT SS mice (Figure 4 right panels). A trend toward less severe glomerular congestion and fibrosis in Fibγ390-396A SS mice compared with FibWT SS mice was also observed (supplemental Figure 6A-B). Tubular damage is also a common characteristic of sickle kidney17 and indeed, tubular damage was apparent in both FibWT SS and Fibγ390-396A SS mice but there was no appreciable improvement of tubular pathology in Fibγ390-396A SS mice (supplemental Figure 7A). Hemosiderin deposition in tubules were similar in FibWT SS and Fibγ390-396A SS mice (supplemental Figure 7B). Taken together, these findings of unattenuated tubular pathology in Fibγ390-396A SS compared with FibWT SS mice stands in contrast to the relative rescue of glomerular damage in the Fibγ390-396A SS mice. Non-sickle, FibWT AA/B6 and Fibγ390-396A AA/B6 mice showed normal kidney histology, including normal glomeruli and tubules (supplemental Figure 8A-B). However, glomerular basement membranes were mildly thickened in 10% to 20% of mice, a finding common in irradiated and older mice.
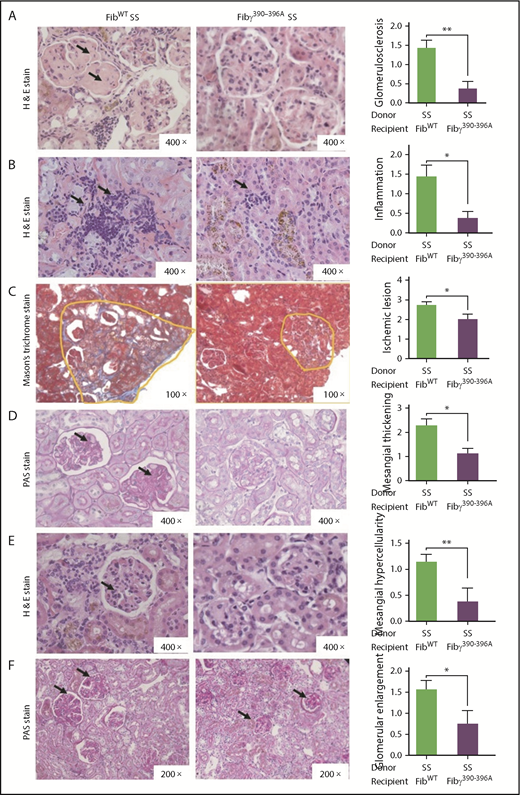
Fibγ390-396Aprotects SCA-associated glomerular pathology. Fully chimeric sickle mice were followed for 1 year posttransplant; kidney samples were fixed in formalin after the mice were euthanized, and stained with H&E and PAS reagent. Representative kidney sections of FibWT SS mice (left panel), Fibγ390-396A SS mice (middle panel) and semiquantitative scoring of histologic features (right panel) showing glomerulosclerosis (A), inflammatory cell infiltration (B), ischemic lesions (C), glomerular mesangial thickening (D), glomerular mesangial hypercellularity (E), and glomerular enlargement (F) are reduced in Fibγ390-396A SS mice compared with the FibWT SS mice. Histopathology scores ranged from 0 to 5, where 0 represents normal kidney morphology; 1, changes in <20% of glomerular area; 2, 21% to 40% of the glomerular area; 3, 41% to 60% of the glomerular area; 4, 61% to 80% of glomerular area; and 5, severe changes in almost all glomeruli. Similar number of fully chimeric male and female mice were used in the experimental groups. The bars in the graph indicate the mean ± SEM. Statistical analyses of the histologic scores were done by Mann-Whitney U test; statistical significance between FibWT SS (n = 7) and Fibγ390-396A SS (n = 8) mice is indicated as **P ≤ .01, *P ≤ .05.
Fibγ390-396Aprotects SCA-associated glomerular pathology. Fully chimeric sickle mice were followed for 1 year posttransplant; kidney samples were fixed in formalin after the mice were euthanized, and stained with H&E and PAS reagent. Representative kidney sections of FibWT SS mice (left panel), Fibγ390-396A SS mice (middle panel) and semiquantitative scoring of histologic features (right panel) showing glomerulosclerosis (A), inflammatory cell infiltration (B), ischemic lesions (C), glomerular mesangial thickening (D), glomerular mesangial hypercellularity (E), and glomerular enlargement (F) are reduced in Fibγ390-396A SS mice compared with the FibWT SS mice. Histopathology scores ranged from 0 to 5, where 0 represents normal kidney morphology; 1, changes in <20% of glomerular area; 2, 21% to 40% of the glomerular area; 3, 41% to 60% of the glomerular area; 4, 61% to 80% of glomerular area; and 5, severe changes in almost all glomeruli. Similar number of fully chimeric male and female mice were used in the experimental groups. The bars in the graph indicate the mean ± SEM. Statistical analyses of the histologic scores were done by Mann-Whitney U test; statistical significance between FibWT SS (n = 7) and Fibγ390-396A SS (n = 8) mice is indicated as **P ≤ .01, *P ≤ .05.
To determine whether increased kidney damage was accompanied by progressive decreased in renal function, we analyzed urine albumin at 6, 9, and 12 months post-BMT and did not observe any significant alteration of urine albumin excretion between FibWT SS vs Fibγ390-396A SS mice at 6 months post-BMT (supplemental Figure 9A), a trend toward reduction of albuminuria at 9 months post-BMT (supplemental Figure 10A), and significant difference in albuminuria only at 12 months post-BMT (Figure 5A), indicating improved glomerular function. At this 12 month time point, the hyperfiltration, reflected in the 24-hour urine volume in sickle mice compared with the nonsickle mice, was also decreased in Fibγ390-396A SS mice compared with FibWT SS mice (Figure 5B). However, we did not observe any significant difference in urine volume of sickle vs nonsickle mice at 6 and 9 months post-BMT (supplemental Figures 9B and 10B).
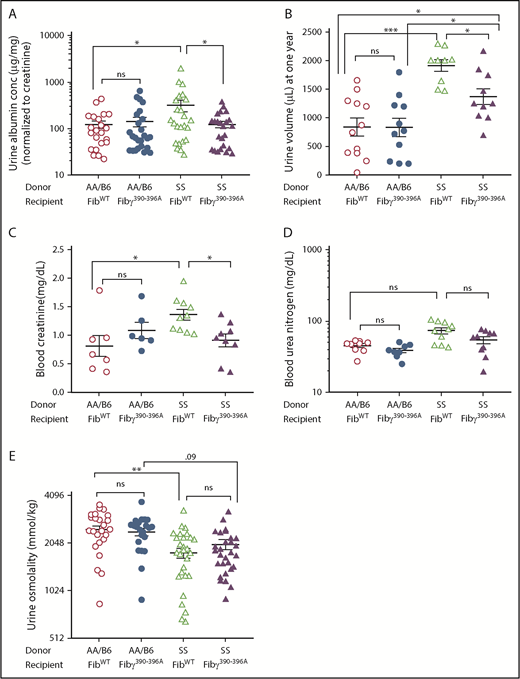
Urine albumin, urine volume, and blood creatinine are reduced in Fibγ390-396ASS mice compared with the FibWTSS mice. One year after BMT, urine samples were collected for 24 hours from the experimental mice and albumin concentration was measured by the mouse albumin ELISA kit. (A) Urine albumin (normalized by urine creatinine) is reduced in Fibγ390-396A SS mice compared with the FibWT SS mice. (B) Urine volume is decreased in Fibγ390-396A SS mice compared with the FibWT SS mice. One year after BMT, blood samples were collected from the experimental mice. Platelet-poor plasma samples were used for urea nitrogen and creatinine measurement. (C) Blood creatinine is decreased in Fibγ390-396A SS mice compared with the FibWT SS mice. (D) Blood urea nitrogen shows a trend for reduction in Fibγ390-396A SS mice compared with the FibWT SS mice. (E) Urine osmolality is not improved in Fibγ390-396A SS mice compared with the FibWT SS mice. Similar number of fully chimeric male and female mice were used in the experimental groups. Each symbol represents an individual mouse. The bars in the graph indicate the mean ± SEM. Statistical significance was determined using 1-way ANOVA followed by the Tukey's multiple comparison test; statistical significance is indicated as ***P ≤ .001, **P ≤ .01, *P ≤ .05. (D) For blood urea nitrogen, the Dunn test were used.
Urine albumin, urine volume, and blood creatinine are reduced in Fibγ390-396ASS mice compared with the FibWTSS mice. One year after BMT, urine samples were collected for 24 hours from the experimental mice and albumin concentration was measured by the mouse albumin ELISA kit. (A) Urine albumin (normalized by urine creatinine) is reduced in Fibγ390-396A SS mice compared with the FibWT SS mice. (B) Urine volume is decreased in Fibγ390-396A SS mice compared with the FibWT SS mice. One year after BMT, blood samples were collected from the experimental mice. Platelet-poor plasma samples were used for urea nitrogen and creatinine measurement. (C) Blood creatinine is decreased in Fibγ390-396A SS mice compared with the FibWT SS mice. (D) Blood urea nitrogen shows a trend for reduction in Fibγ390-396A SS mice compared with the FibWT SS mice. (E) Urine osmolality is not improved in Fibγ390-396A SS mice compared with the FibWT SS mice. Similar number of fully chimeric male and female mice were used in the experimental groups. Each symbol represents an individual mouse. The bars in the graph indicate the mean ± SEM. Statistical significance was determined using 1-way ANOVA followed by the Tukey's multiple comparison test; statistical significance is indicated as ***P ≤ .001, **P ≤ .01, *P ≤ .05. (D) For blood urea nitrogen, the Dunn test were used.
Furthermore, blood creatinine were increased in FibWT SS mice compared with FibWT AA/B6 mice and was decreased in Fibγ390-396A SS mice compared with the FibWT SS mice (Figure 5C), indicating improved renal function in Fibγ390-396A SS mice. Blood urea nitrogen showed a trend for increase in FibWT SS mice compared with FibWT AA/B6 mice and a trend for decrease in Fibγ390-396A SS mice compared with the FibWT SS mice (Figure 5D).
We also assessed urine concentration ability, loss of which develops early in mice43 and humans with SCA and is largely irreversible after adolescence.44 Both FibWT SS and Fibγ390-396A SS mice had significantly diminished urine osmolality compared with nonsickle mice (Figure 5E). Urine osmolality was not significantly different between the FibWT SS and Fibγ390-396A SS mice at 1 year post-BMT (Figure 5E), consistent with no improvement of tubular pathology in Fibγ390-396A mice. Taken together, these data demonstrate that imposition of Fibγ390-396A mutation offers significant protection from development of glomerular pathology and downstream albuminuria associated with SCA but does not affect urine concentration ability.
Male mice tended to have slightly higher mortality than female mice (38.5% vs 36.1%) although these differences were not significant (supplemental Table 2). We also found slightly higher (albeit not statistically significant) mortality in FibWT SS mice compared with the Fibγ390-396A SS mice (39.3% vs 34.4%), which suggests that the Fibγ390-396A mutation may provide a subtle survival benefit (supplemental Table 2). We did not observe any significant difference in renal histopathologic features in male vs female SS mice (supplemental Figure 11).
To determine whether the fibrinogen γ390-396 motif is a determinant of sickle nephropathy, analyses of fibrin deposition were performed and indicated that FibWT SS and Fibγ390-396A SS mice showed a similar level of fibrin deposition (supplemental Figure 12A). We did not find any significant increase of plasma fibrinogen in FibWT SS mice compared with nonsickle mice. However, we found significantly reduced plasma fibrinogen in Fibγ390-396A SS than FibWT SS mice (supplemental Figure 13), indicating reduced inflammation in Fibγ390-396A SS mice because fibrinogen is an acute-phase reactant that increases with inflammation. However, consistent with reduced circulating proinflammatory mediators, in SS mice with the mutant Fibγ390-396A, a trend toward reduction of renal macrophages was observed in Fibγ390-396A SS mice compared with FibWT SS mice (supplemental Figure 12B).
As a measure of thrombin generation, we compared plasma thrombin-antithrombin complex (TAT) levels across each cohort. Paralleling previous results,17 there was no significant difference in plasma TAT in the FibWT SS mice relative to FIbWT AA/B6 mice. There were also no significant differences in TAT levels between FibWT SS and Fibγ390-3906A SS mice (supplemental Figure 13B), consistent with the fact that the Fibγ390-3906A mutation would not be expected to alter thrombin generation potential. As a measure of fibrin(ogen) degradation, we analyzed the D-dimers of the plasma of our experimental mice. We did not observe a reduction of D-dimers in Fibγ390-396A AA/B6 mice compared with FibWT AA/B6 mice. We also did not observe a reduction of D-dimers in Fibγ390-396A SS mice compared with FibWT SS mice (supplemental Figure 13C), which is in line with the fibrin deposition in the kidney sections.
We next performed transcriptome profiling of FibWT SS and Fibγ390-396A SS glomeruli, as well as corresponding FibWT B6 and Fibγ390-396A B6 mice to identify differentially expressed genes associated with genetic and glomerular histopathology patterns, and whose pathway-associated functions could reveal additional insights. Glomerular transcriptome of SS mice showed a higher proportion of downregulated gene expression relative to the number of upregulated genes as compared with WT controls (816 vs 271; Figure 6; supplemental Figure 14; supplemental Table 3 clusters 4-8 vs 1-3).
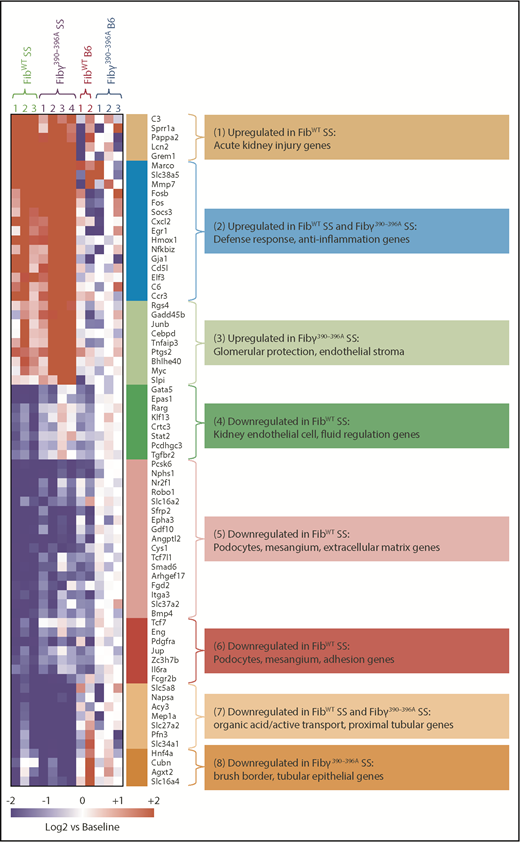
Glomerular injury responses to SCA are strongly modified by the Fib γ390-396domain. RNA-sequencing analyses of glomerular fractions of kidney from the experimental mice 1 year posttransplant reveal chronic injury and loss of normal cellular programming in FibWT SS mice compared with the Fibγ390-396A SS mice. Heatmap data and gene-ontology pathways showing differentially expressed transcripts in FibWT SS (n = 3) vs Fibγ390-396A SS (n = 4) mice glomeruli. FibWT B6 (n = 2) and Fibγ390-396A B6 (n = 3) mice glomeruli were used as control. The most significant enrichments among each of the clusters are indicated by color codes (ToppGene functional enrichments were determined using the complete clusters as shown in supplemental Table 3) and indicate perturbation of the biological processes in the FibWT SS and/or the Fibγ390-396A SS group. Also see supplemental Figure 14 as well as supplemental Table 3 for expanded gene lists associated with each gene cluster. Differentially expressed genes were determined based on Welch ANOVA using a false discovery rate of 0.1 and fold-change >2 for any group comparison.
Glomerular injury responses to SCA are strongly modified by the Fib γ390-396domain. RNA-sequencing analyses of glomerular fractions of kidney from the experimental mice 1 year posttransplant reveal chronic injury and loss of normal cellular programming in FibWT SS mice compared with the Fibγ390-396A SS mice. Heatmap data and gene-ontology pathways showing differentially expressed transcripts in FibWT SS (n = 3) vs Fibγ390-396A SS (n = 4) mice glomeruli. FibWT B6 (n = 2) and Fibγ390-396A B6 (n = 3) mice glomeruli were used as control. The most significant enrichments among each of the clusters are indicated by color codes (ToppGene functional enrichments were determined using the complete clusters as shown in supplemental Table 3) and indicate perturbation of the biological processes in the FibWT SS and/or the Fibγ390-396A SS group. Also see supplemental Figure 14 as well as supplemental Table 3 for expanded gene lists associated with each gene cluster. Differentially expressed genes were determined based on Welch ANOVA using a false discovery rate of 0.1 and fold-change >2 for any group comparison.
Cluster 1 was a group of genes elevated in FibWT SS glomeruli that consisted of genes known to be associated with acute kidney injury, and myeloid and stromal responses. Cluster 2 consisted of genes whose expression was elevated in both FibWT SS and Fibγ390-396A SS, characterized by anti-inflammatory function. Cluster 3 consisted of genes that were significantly overexpressed in Fibγ390-396A SS relative to FibWT SS and consisted of genes associated with a protective glomerular injury response.
Downregulated genes were represented in 3 separate gene clusters (clusters 4, 5, and 6). Genes significantly downregulated in FibWT SS glomeruli relative to other groups consisted of genes characterized by podocyte and mesangial cell signatures as well as genes associated with cell adhesion and endothelium. The final signatures/clusters 7 and 8 consisted of genes downregulated in both FibWT SS and Fibγ390-396A SS relative to B6 mice or just in Fibγ390-396A SS, respectively. These signatures were associated with organic acid/active transport and proximal tubules (group 7), and brush border-critical genes (group 8).
Taken together, these results suggest that Fibγ390-396A strongly modifies cell type–specific response of the glomeruli to the SS model by lessening the acute injury and inflammatory response, improving the protective responses, and preventing the loss of podocytes, mesangium, and endothelial cells.
Based upon the loss of podocyte signature in transcriptome, we validated podocyte structure by immunofluorescence staining of kidney sections with a podocyte marker, Wilms tumor 1 (WT1). Figure 7A demonstrates typical WT1 expression in podocytes of FibWT B6 and Fibγ390-396A B6 mice. In contrast, WT1 expression was significantly reduced in FibWT SS glomeruli (Figure 7B), indicating podocyte damage, whereas Fibγ390-396A SS mice kidney sections showed a WT1 expression pattern more consistent with nonsickle B6 mice, suggesting protection of podocytes in Fibγ390-396A SS glomeruli (Figure 7B), in line with the glomerular transcriptome. However, how the Fibγ390-396A domain of fibrinogen protects against podocyte loss remains to be studied.
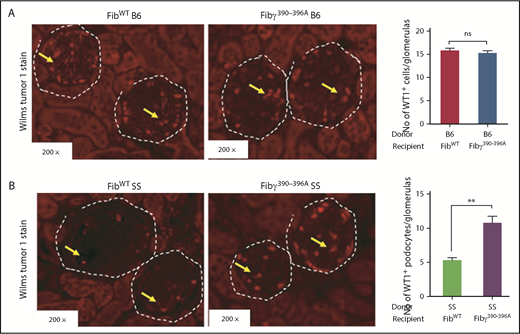
Immunofluorescence staining reveals reduced expression of podocyte marker, WT1 in FibWTSS compared with the Fibγ390-396ASS mice. Fully chimeric similar number of male and female mice were followed for 1 year posttransplant and kidney samples were fixed in formalin after the mice were euthanized, and stained with anti-WT1 antibody. The number of WT1+ podocytes were counted from 20 glomeruli of each kidney section and average number of WT1+ podocytes per glomerulus were calculated. (A) Kidney sections showing similar level of WT1 expression (inside the white dotted circle) in podocytes of glomeruli from FibWT B6 (left panel, n = 6) and Fibγ390-396A B6 (right panel, n = 6) mice. (B) WT1 expression (inside the white dotted circle) is depleted in podocytes of glomeruli of FibWT SS (n = 6) mice compared with the Fibγ390-396A SS (n = 6) mice. The bars in the graph indicate the mean ± SEM. Statistical analyses were done by Mann-Whitney U test and statistical significance are indicated as **P ≤ .01.
Immunofluorescence staining reveals reduced expression of podocyte marker, WT1 in FibWTSS compared with the Fibγ390-396ASS mice. Fully chimeric similar number of male and female mice were followed for 1 year posttransplant and kidney samples were fixed in formalin after the mice were euthanized, and stained with anti-WT1 antibody. The number of WT1+ podocytes were counted from 20 glomeruli of each kidney section and average number of WT1+ podocytes per glomerulus were calculated. (A) Kidney sections showing similar level of WT1 expression (inside the white dotted circle) in podocytes of glomeruli from FibWT B6 (left panel, n = 6) and Fibγ390-396A B6 (right panel, n = 6) mice. (B) WT1 expression (inside the white dotted circle) is depleted in podocytes of glomeruli of FibWT SS (n = 6) mice compared with the Fibγ390-396A SS (n = 6) mice. The bars in the graph indicate the mean ± SEM. Statistical analyses were done by Mann-Whitney U test and statistical significance are indicated as **P ≤ .01.
Discussion
Previous studies have shown that coagulation proteases (eg, factor Xa and thrombin) are linked to the pathologies of SCA.17,45 Indeed, genetic or pharmacological reduction of circulating prothrombin was shown to be protective against SCA-associated inflammation, end-organ damage, and mortality in mice.17,45 The fact that reduction in thrombin generation results in improved SCA associated multiorgan pathologies is not surprising because thrombin has numerous downstream pathways.19,46 Here, we evaluated the possibility that fibrinogen is a thrombin downstream target mediating development of SCA-associated end-organ pathologies. The studies here demonstrate that interaction of the γ390-396 motif of fibrinogen with leukocytes drives WBC ROS production, systemic inflammation, and development of kidney glomerular pathologies and dysfunction.
Chen et al reported improved blood flow rate and prolonged survival in experimentally induced acute vaso-occlusive crisis sickle mice deficient in Mac-1 (αMβ2) integrin (the domain that binds Fibγ390-396), but showed no improvement in chronic organ damage at 6 months posttransplant.47 To study organ damage, Chen et al transplanted sickle cell disease mice deficient in Mac-1 (SCD.Mac-1−/−) bone marrow into C57BL/6 recipients. Here, the effect of Mac-1 deletion in reducing SCA-associated end-organ damage would not develop by 6 months after SS transplant because recipients have healthy organs for 3 months before full sickle RBC chimerism develops, due to the extremely short 1- to 2-day lifespan of SS RBCs as compared with normal mouse RBCs.48,49 We have shown that it takes 6 to 9 months after complete RBC chimerism for normal recipient organs to develop SCA-associated pathology.17,43,50 In native SS mice (nontransplanted), organ damage appears at 4 to 6 months of age48 because the mice switch to sickle hemoglobin ensues early. To see a difference in organ pathology between sickle mice with normal fibrinogen and Fibγ390-396A, we needed to follow for 9 months after they attained full sickle RBC chimerism (total 1 year follow up after BMT). In contrast, in the study by Chen et al, the mice were analyzed 6 months after transplant, which is ∼3 months after attaining full sickle RBC chimerism; they have not seen significant organ damage.
We did not observe reduction of albuminuria in Fibγ390-396A SS mice at 6 months, a trend toward reduction at 9 months, and significantly reduced albuminuria at 12 months post-BMT, compared with FibWT SS mice. The major difference in our study is remarkably improved glomerular pathology in Fibγ390-396A SS mice, which was not seen in SCD.Mac-1−/− mice likely from the shorter duration of SCA in their study.47 Also, the γ390-396 of fibrinogen mediates FXIII binding and activation,51 which may play a role in sickle glomerular pathology, and it is possible that Fibγ390-396 binding to FXIII may augment inflammatory cell recruitment to the kidney.
Sickle cell nephropathy is a well-known cause of morbidity and mortality of SCA, and the incidence of renal complications increases with long-term survival of patients.41,52 The underlying relationship of sickle cell nephropathy, renal insufficiency, and glomerular lesions to SCA is poorly understood. SCA patients show renal histopathologic features, including thickening of the cortex, glomerular enlargement, and mesangial proliferation, followed by fibrosis, focal segmental glomerulosclerosis, besides renal congestion, tubulointerstitial inflammation, and tubular iron deposition.53-56 In our study, fibrinogen binding to αMβ2 on leukocytes appeared to be essential in SCA-related glomerular pathology such that interruption of fibrinogen binding in Fibγ390-396A SS mice results in a striking reduction of glomerulosclerosis, inflammatory cell infiltration, ischemic lesions, mesangial thickening, mesangial hypercellularity, and glomerular enlargement compared with FibWT SS mice.
Interestingly, Fibγ390-396A only provides beneficial effects in kidney pathology of SS mice but not in other organs. The major difference noted upon extensive analysis was a significant reduction of inflammatory infiltrates and presence of activated macrophages in the Fibγ390-396A SS kidney. However, we did not find reduction of inflammatory infiltrates in other organs. Downregulation of podocyte and mesangial signatures in our FibWT SS mice, with loss of podocytes in immunohistology, whereas upregulation of genes for glomerular protection in Fibγ390-396A SS mice provide an explanation for reduced albuminuria in Fibγ390-396A SS mice compared with FibWT SS mice. Fibrinogen induces podocyte damage via Toll-like receptor 4 through the p38 MAPK and NF-κB p65 pathway57 or in diabetic glomerulopathy; increased extracellular glucose-mediated ROS generation led to the activation of proapoptotic p38 MAPK and caspase 3 and causes apoptosis of podocytes.58 We observed reduced ROS production in WBCs and decreased numbers of activated macrophages in the Fibγ390-396A SS kidney that may provide glomerular protective responses in Fibγ390-396A SS mice that have been studied.
We thoroughly analyzed the histology slides of the organ samples and did not observe any definitive differences of microthrombi size in FibWT SS vs Fibγ390-396A SS mice. It may be that microthrombi were not well captured in the histology sections to comment on the size differences. In addition, immunohistochemistry staining of fibrin showed a similar pattern in both groups where no microthrombi was obviously highlighted. It is still possible, of course, that differences are not seen in this histology. Therefore, the reduced glomerular injury might be due to fewer microthrombi in addition to reduced inflammation.
A careful assessment of cardiac phenotype demonstrates a subtle cardiac benefit in Fibγ390-396A SS mice. This finding may be due to improved renal status in Fibγ390-396A SS mice, with potentially less activation of the renin-angiotensin axis. Interestingly, IL-6 has been reported to mediate angiotensin II–induced myocardial fibrosis, cardiac hypertrophy, and diastolic dysfunction.59,60 Although in our previous study,17 we found a large reduction in plasma IL-6 levels in SS mice with low prothrombin compared with SS mice with normal prothrombin levels, which correlated with improved cardiac pathology in the former study. In our current study, the reduction of IL-6 levels in Fibγ390-396A SS mice compared with FibWT SS mice was modest, and this correlates with the subtle improvement in cardiac function. Interestingly, cardiac phenotype in FibWT SS mice differs from our previous study in Berkeley sickle mice.50 This may be due to inherent differences between 2 models: our previous study used native Berkeley sickle mice50 whereas in our current study, we transplanted Berkeley sickle bone marrow into lethally irradiated mice to induce SCA phenotype.50 It could also be due to survival of the “fitter” SCA mice. We experienced attrition within the FibWT SS and Fibγ390-396A SS cohorts over the course of our studies, and indeed our studies of the Berkley sickle mice were conducted at 8 months of age. Thus, examining the mice at 1 year posttransplant may have resulted in a survivorship phenomenon, with more severely SCA affected mice having died earlier leaving a relatively “healthier” cohort for study at the older time point.
In summary, our study demonstrates that fibrinogen binding to αMβ2 integrin on leukocytes specifically causes glomerular injury due to the increased leukocyte ROS production and inflammation, resulting in podocyte loss and glomerulosclerosis in a mouse model of SCA. Based on these findings, it will be interesting to decipher the fibrinogen-specific signaling pathways that are activated in SCA and test the targeted therapeutic options to inhibit fibrinogen binding to αMβ2 or its downstream signaling partners, which may provide beneficial effects in kidney disease of SCA.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Matthew J. Flick (Cincinnati Children’s Hospital Medical Center) for generous provision of the Fibγ390-396A and FibWT mice, input in research, and critical review of the manuscript; Anastacia Loberg (Cincinnati Children’s Hospital Medical Center) for technical help in animal works; and the Flow Cytometry Core, Veterinary Core, and Pathology Core of Cincinnati Children’s Hospital Medical Center for their services.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL112603 (P.M.) and start-up funding from Cincinnati Children’s Research Foundation (M.N.).
Authorship
Contribution: M.N., P.I.A., and E.S.M. performed the experiments; M.N. analyzed and plotted the data; M.N., P.M., P.I.A., and E.S.M. designed experiments and interpreted the data; M.G.N. and M.A.S. did the immunohistochemistry and immunofluorescence staining; S.M. helped in mouse breeding, bone marrow transplants, and experimental procedures; J.M.J. performed the cardiac imaging analyses; K.V. performed the organ histopathology analysis and scoring; B.J.A. performed the RNA-sequencing analyses and interpretation; M.N. and P.M. wrote the manuscript; and all authors read the manuscript and provided their comments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Md Nasimuzzaman, Cincinnati Children's Medical Center, 240 Albert Sabin Way, Mail Code #7013, Cincinnati, OH 45229; e-mail: md.nasimuzzaman@cchmc.org or mdnasimuzzaman@yahoo.com.