

Key Points
This article characterizes a cryptic IGH/CCND1 rearrangement in MCL by NGS.
Mate-pair sequencing can help in accurately diagnosing MCL in cases of cyclin-D1–positive B-cell lymphoma with negative CCND1 FISH studies.
Introduction
Mantle cell lymphoma (MCL) is a mature B-cell neoplasm accounting for approximately 3% to 10% of all non-Hodgkin lymphomas.1-3 The molecular hallmark of MCL, t(11;14)(q13;q32) (IGH/CCND1), results in deregulation and overexpression of the G1-phase cell cycle gene, CCND1 (encoding cyclin D1), mediated by enhancer elements found throughout the IGH locus.1-4 The t(11;14) is observed in >95% of MCL cases, with rare reports that describe t(11;14) as being a result of CCND1 and immunoglobulin light chain translocations.1 Cyclin D1–negative MCL resulting from CCND2 or CCND3 rearrangements and immunoglobulin partners highlights the importance of cyclin D overexpression in all MCL subtypes.1,3-6 However, overexpression of cyclin D alone is insufficient to cause MCL, and additional genetic alterations that deregulate cell cycle, DNA damage response, apoptosis, and/or chromatin modifiers are typically required.1-3,7-12 Although cyclin D1 overexpression can occur in other mature B-cell lymphomas such as hairy cell leukemia and rarely chronic lymphocytic leukemia/small lymphocytic lymphoma, IGH/CCND1 fusion detected in mature B-cell lymphomas has been considered diagnostic for MCL.1,13,14
IGH/CCND1 rearrangements are typically detected by fluorescence in situ hybridization (FISH) studies using dual-color dual-fusion (D-FISH) or less commonly by CCND1 break-apart probe sets. We describe a unique case of cyclin D1–positive B-cell lymphoma with partial features of MCL but without CCND1 rearrangement by commercially available FISH probe sets. To further interrogate this discrepancy, a next-generation sequencing (NGS) strategy, mate-pair sequencing (MPseq), was performed, and it revealed a cryptic IGH/CCND1 rearrangement.
Case description
A 79-year-old woman presented with fatigue, decreased appetite, and dizziness of 2-week duration. Her hematologic laboratory profile was significant for a total white blood cell (WBC) count of 94.8 × 109/L (95% lymphocytes), hemoglobin 10.1 g/dL, and platelets 143 × 109/L. A computed tomography scan of chest, abdomen, and pelvis and positron emission tomography scan demonstrated generalized lymphadenopathy and splenomegaly. FISH studies revealed TP53 deletion but were negative for IGH/CCND1 fusion and CCND2 rearrangement. CCND3 rearrangement studies were not performed. Hematopathologic review demonstrated strong, uniform overexpression of cyclin D1, positivity for CD5 and a lack of CD200 expression by the neoplastic lymphocytes, which support a diagnosis of MCL. The lack of SOX11 expression by the lymphoma cells is highly uncommon in MCL, although it can occur in indolent or TP53 mutation–associated MCL.15,16 Because cyclin D1 overexpression by lymphoma cells is not 100% specific for MCL in the absence of IGH/CCND1 fusion, an initial diagnosis of cyclin D1–positive B-cell lymphoma was established, and the patient was subsequently treated with ibrutinib.
After 18 months of ibrutinib therapy with partial remission, the patient progressed with a rapidly rising WBC count (119.5 × 109/L; 89% lymphocytes). No evidence of acquired BTK or PLCG2 mutations were detected in the peripheral blood. The peripheral blood smear revealed marked lymphocytosis with predominantly small lymphocytes (85% of total cells) with condensed chromatin, scant cytoplasm, and prominent cytoplasmic vacuoles (Figure 1A-B). The bone marrow was hypercellular (approximately 70%) with 70% to 80% involvement by the mature lymphocytes (Figure 1C-D). The lymphocytes were CD20+, coexpressed cyclin D1, lacked expression for SOX11 (Figure 1E-G), and also lacked hairy cell leukemia–associated markers tartrate-resistant acid phosphatase, DBA4.4, T-bet, and annexin A1 (data not shown). Flow cytometry revealed λ light chain–restricted B lymphocytes, positive for CD20 and CD19, partially positive for CD5, and negative for CD200 (Figure 1H-I). FISH analysis revealed TP53 and 13q deletions, gain of BCL3, and an extra IGH signal in ∼59% to 77% of interphase nuclei (supplemental Table 1). The IGH/CCND1 D-FISH and CCND1 break-apart probe studies were negative (Figure 2A-B). MPseq and a 61-gene NGS panel were subsequently performed on the bone marrow specimen.
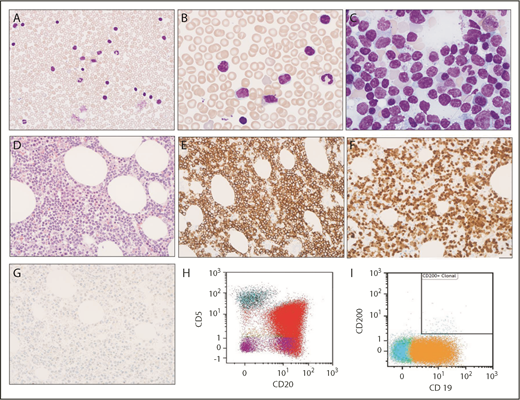
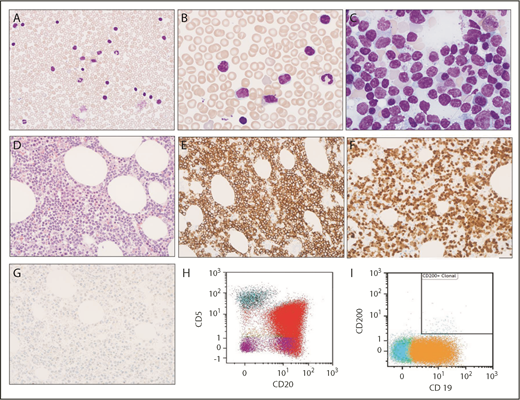
Morphologic and immunophenotypic evaluation. (A-B) Low (original magnification ×400) and high (original magnification ×1000) magnification of Wright-Giemsa stained peripheral blood smear show circulating small lymphocytes, some of which contain cytoplasmic vacuoles. (C) Wright-Giemsa stained bone marrow aspirate smear at high (original magnification ×1000) magnification shows similar small lymphocytes with cytoplasmic vacuoles. (D) Hematoxylin and eosin–stained bone marrow biopsy (original magnification ×400) shows extensive infiltration with small lymphocytes. (E-G) Immunohistochemical stains indicate that lymphocytes are positive for CD20 (E) and cyclin D1 (F) but are negative for SOX11 (G). (H-I) Immunophenotyping by flow cytometry demonstrates λ light chain–restricted B lymphocytes are positive for CD20 (red) and CD19 (orange), partially positive for CD5, and negative for CD200.
Morphologic and immunophenotypic evaluation. (A-B) Low (original magnification ×400) and high (original magnification ×1000) magnification of Wright-Giemsa stained peripheral blood smear show circulating small lymphocytes, some of which contain cytoplasmic vacuoles. (C) Wright-Giemsa stained bone marrow aspirate smear at high (original magnification ×1000) magnification shows similar small lymphocytes with cytoplasmic vacuoles. (D) Hematoxylin and eosin–stained bone marrow biopsy (original magnification ×400) shows extensive infiltration with small lymphocytes. (E-G) Immunohistochemical stains indicate that lymphocytes are positive for CD20 (E) and cyclin D1 (F) but are negative for SOX11 (G). (H-I) Immunophenotyping by flow cytometry demonstrates λ light chain–restricted B lymphocytes are positive for CD20 (red) and CD19 (orange), partially positive for CD5, and negative for CD200.
![Figure 2. Cytogenomic evaluation. (A) Representative interphase cell demonstrating 2 red signals (CCND1 probe, ∼378 kb) and 3 green signals (IGH probe, ∼1.6 Mb) using an IGH/CCND1 dual-color dual-fusion probe set. This signal pattern indicates a gain of 14q32 or an IGH rearrangement, without the presence of IGH/CCND1 fusion. The presence of an unbalanced or balanced t(11;14) would be indicated by 1 or 2 red/green (yellow) fusion signals, respectively. (B) Representative interphase cell demonstrating 2 intact yellow fusion signals (5′ CCND1 probe, ∼384 kb [green]; 3′ CCND1 probe, ∼256 kb [red]) using a CCND1 break-apart probe (BAP) set. The presence of a CCND1 rearrangement would be indicated by separated or single green (5′ CCND1 probe) and red (3′ CCND1 probe) signals. (C) Junction plot demonstrating the insertion of an intact CCND1 gene into the IGH locus. In addition, loss of a portion of distal IGH-J, IGH-D, and a portion of the proximal IGH-V region was detected (2N indicated by the dashed gray horizontal line below the IGH locus label). (D) A depiction of the cryptic CCND1 insertional event. Dashed horizontal red lines on the normal chromosome 11 indicate breaks encompassing the CCND1 gene and insertion into the IGH locus located on the der(14). The black dots on IGH locus indicate enhancer elements. The IGH segment between the horizontal dashed red lines on chromosome 14 was deleted. The IGH/CCND1 D-FISH and CCND1 BAP footprints are indicated by the solid green/red and striped green/red horizontal bars, respectively. Insertion of the CCND1 gene into the IGH locus is unappreciable by FISH because of the minimal size of the inserted CCND1 gene segment. The secondary 12p/der(14q) translocation (not shown) also accounts for the extra IGH signal observed by IGH/CCND1 D-FISH studies.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/8/10.1182_bloodadvances.2019031450/2/m_advances031450f2.png?Expires=1768774386&Signature=OVRkAbHHrsidGbT~W4HedWwzRsGLaBtWL5mPZg-rQh45gu7CQLZtRRY7oSsvEnev3sLRXMHLpf3F0czrctIacstegH~nvcEZMdF3BDpuPNvz0AKZ9gdOs-o~wEeL1Rox0W~ainvoIp~k1nLzfsGKhCumHfwHl9~sCm9pJbA7cgHeX-rH-BsUsnFVTqxS9lU3WHTwfq7LnFAepFnf917ORdQcdyf8W~SU6inScaIrea~kh~a4upz~dApY4uLSLdplybLvWwLMvTBJk4L~dfbRju8Q8xjfYdpkCpInNaExNNjkQuVFP0orGEuSXvFY41L0-1BCsv82bIoKefe-emL22Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cytogenomic evaluation. (A) Representative interphase cell demonstrating 2 red signals (CCND1 probe, ∼378 kb) and 3 green signals (IGH probe, ∼1.6 Mb) using an IGH/CCND1 dual-color dual-fusion probe set. This signal pattern indicates a gain of 14q32 or an IGH rearrangement, without the presence of IGH/CCND1 fusion. The presence of an unbalanced or balanced t(11;14) would be indicated by 1 or 2 red/green (yellow) fusion signals, respectively. (B) Representative interphase cell demonstrating 2 intact yellow fusion signals (5′ CCND1 probe, ∼384 kb [green]; 3′ CCND1 probe, ∼256 kb [red]) using a CCND1 break-apart probe (BAP) set. The presence of a CCND1 rearrangement would be indicated by separated or single green (5′ CCND1 probe) and red (3′ CCND1 probe) signals. (C) Junction plot demonstrating the insertion of an intact CCND1 gene into the IGH locus. In addition, loss of a portion of distal IGH-J, IGH-D, and a portion of the proximal IGH-V region was detected (2N indicated by the dashed gray horizontal line below the IGH locus label). (D) A depiction of the cryptic CCND1 insertional event. Dashed horizontal red lines on the normal chromosome 11 indicate breaks encompassing the CCND1 gene and insertion into the IGH locus located on the der(14). The black dots on IGH locus indicate enhancer elements. The IGH segment between the horizontal dashed red lines on chromosome 14 was deleted. The IGH/CCND1 D-FISH and CCND1 BAP footprints are indicated by the solid green/red and striped green/red horizontal bars, respectively. Insertion of the CCND1 gene into the IGH locus is unappreciable by FISH because of the minimal size of the inserted CCND1 gene segment. The secondary 12p/der(14q) translocation (not shown) also accounts for the extra IGH signal observed by IGH/CCND1 D-FISH studies.
Cytogenomic evaluation. (A) Representative interphase cell demonstrating 2 red signals (CCND1 probe, ∼378 kb) and 3 green signals (IGH probe, ∼1.6 Mb) using an IGH/CCND1 dual-color dual-fusion probe set. This signal pattern indicates a gain of 14q32 or an IGH rearrangement, without the presence of IGH/CCND1 fusion. The presence of an unbalanced or balanced t(11;14) would be indicated by 1 or 2 red/green (yellow) fusion signals, respectively. (B) Representative interphase cell demonstrating 2 intact yellow fusion signals (5′ CCND1 probe, ∼384 kb [green]; 3′ CCND1 probe, ∼256 kb [red]) using a CCND1 break-apart probe (BAP) set. The presence of a CCND1 rearrangement would be indicated by separated or single green (5′ CCND1 probe) and red (3′ CCND1 probe) signals. (C) Junction plot demonstrating the insertion of an intact CCND1 gene into the IGH locus. In addition, loss of a portion of distal IGH-J, IGH-D, and a portion of the proximal IGH-V region was detected (2N indicated by the dashed gray horizontal line below the IGH locus label). (D) A depiction of the cryptic CCND1 insertional event. Dashed horizontal red lines on the normal chromosome 11 indicate breaks encompassing the CCND1 gene and insertion into the IGH locus located on the der(14). The black dots on IGH locus indicate enhancer elements. The IGH segment between the horizontal dashed red lines on chromosome 14 was deleted. The IGH/CCND1 D-FISH and CCND1 BAP footprints are indicated by the solid green/red and striped green/red horizontal bars, respectively. Insertion of the CCND1 gene into the IGH locus is unappreciable by FISH because of the minimal size of the inserted CCND1 gene segment. The secondary 12p/der(14q) translocation (not shown) also accounts for the extra IGH signal observed by IGH/CCND1 D-FISH studies.
Methods
All FISH probes were supplied by Abbott Molecular (Des Plaines, IL) with the exception of our laboratory-developed IGH/BCL3 D-FISH probe set (details provided in supplemental Table 1). For MPseq, DNA was processed using the Illumina Nextera Mate Pair library preparation kit (Illumina, San Diego, CA) and sequenced on the Illumina HiSeq 2500 using 101 base pair reads and paired-end sequencing. Data were aligned to the reference genome (GRCh38) using BIMA V3, and abnormalities were identified and visualized using in-house–developed bioinformatics tools, SVAtools, and in-house–developed visualization tool, Ingenium.17,18 NGS was performed using a 61-gene panel (supplemental Table 2). Approximately 200 ng of genomic DNA sheared to approximately 150 base pairs was used for library preparation by the SureSelect XT library kit (Agilent, Santa Clara, CA) and sequenced on the HiSeq platform (Illumina) using 2 × 101 read lengths. Data were aligned to the reference genome (GRCh38) and processed through an in-house–developed pipeline, Mayo NGS Workbench, for base calling and insertion/deletion (indel) detection. Results were confirmed by reviewing the sequencing files (.bam file format) in Alamut Visual (Interactive Biosoftware, Rouen, France). This study was conducted in accordance with the Declaration of Helsinki.
Results and discussion
The discordant findings of strong cyclin D1 positivity by immunohistochemistry vs the lack of CCND1 rearrangement by FISH were resolved by MPseq results, which detected an insertional rearrangement involving chromosomes 11 and 14. This resulted in an intact coding region of the CCND1 gene (NM_053056) from chromosome 11 inserted into the IGH locus on chromosome 14 (Figure 2C). The insertion of CCND1 into the IGH locus characterized by MPseq (Figure 2D) explains the cyclin D1 overexpression observed by immunohistochemistry (Figure 1F). The identification of cryptic IGH/CCND1 fusion by MPseq was critical in establishing a conclusive diagnosis of MCL in this patient vs the diagnosis of cyclin D1–positive B-cell lymphoma established at presentation. The cryptic nature of this particular genetic alteration eluded detection by standard FISH techniques because of the minimal size of the CCND1 gene segment inserted into the IGH locus (Figure 2D).
A subsequent balanced translocation was identified between 12p11.1 (intergenic region) and the CCND1 3′ untranslated region (UTR) located on the derivative chromosome 14, resulting in a disrupted CCND1 3′ UTR (data not shown). Both rearrangements were confirmed by Sanger sequencing and involved the same CCND1 allele (supplemental Table 3). A retrospective analysis of the diagnostic bone marrow specimen by MPseq detected only the CCND1 insertion event, indicating that the 12p/der(14q) translocation occurred secondarily during clonal evolution. Of interest, the translocation of the CCND1 3′ UTR region and 12p11.2 chromosomal region has been described in cyclin D1–positive MCL without IGH/CCND1 fusion and may indicate a rare but recurrent mechanism for cyclin D1 overexpression.11 Truncation of the CCND1 3′ UTR creates transcripts that lack messenger RNA destabilization elements, thus extending the half-life of the modified messenger RNA transcript and increasing its oncogenic effect.8,9
Targeted panel NGS studies also revealed single nucleotide pathogenic mutations involving CCND1 and TP53 (supplemental Table 4). The combination of cryptic IGH/CCND1 fusion, acquisition of the CCND1 3′ UTR translocation involving 12p in conjunction with biallelic TP53 alterations (deletion and point mutation), and the reported adverse effect of acquired CCND1 mutations in MCL also explain the negative SOX11 expression by the lymphoma cells and may have contributed to progression after an initial response to ibrutinib therapy in this case.10,16 The patient was started on venetoclax, which resulted in a rapid decline of her WBC count, resolution of splenomegaly, and clinical improvement of her symptoms. Unfortunately, the response to venetoclax was not durable, and the patient expired as a result of progression of MCL 2 months later.
In conclusion, we have characterized a novel IGH/CCND1 rearrangement using NGS that was undetectable by FISH and provided definitive evidence for a diagnosis of MCL. The comprehensive genetic characterization of this case revealed additional key findings that seem important in disease progression and therapeutic resistance.
The full-text version of this article contains a data supplement.
Acknowledgment
This work was supported by the Department of Laboratory Medicine and Pathology, Mayo Clinic.
Authorship
Contribution: J.F.P. drafted and edited the manuscript, generated figures, and helped analyze the data; L.B.B. participated in writing and editing the manuscript and helped analyze the data; R.P.K. collected and analyzed data and edited the manuscript; B.A.P. and S.A.S. analyzed and interpreted data, generated figures, and edited the manuscript; G.V., J.B.S., and P.T.G. reviewed and edited the manuscript; A.A.M., C.A.T., and S.A.P. collected clinical data and participated in writing and editing the manuscript; and D.C. and D.S.V. collected and analyzed data, generated figures, and participated in writing and editing the manuscript.
Conflict-of-interest disclosure: Algorithms described in this manuscript for mate-pair sequencing are licensed to WholeGenome LLC owned by G.V. S.A.P. reports research funding to his institution from Pharmacyclics, MorphoSys, Janssen, AstraZeneca, and Ascentage Pharma for clinical studies in which he is a principal investigator. S.A.P. has also participated in Advisory Board meetings of Pharmacyclics, AstraZeneca, Genentech, Gilead, and AbbVie (not personally compensated for his participation). The remaining authors declare no competing financial interests.
Correspondence: Linda B. Baughn, Division of Laboratory Genetics and Genomics, Department of Laboratory Medicine and Pathology, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: baughn.linda@mayo.edu.

![Figure 2. Cytogenomic evaluation. (A) Representative interphase cell demonstrating 2 red signals (CCND1 probe, ∼378 kb) and 3 green signals (IGH probe, ∼1.6 Mb) using an IGH/CCND1 dual-color dual-fusion probe set. This signal pattern indicates a gain of 14q32 or an IGH rearrangement, without the presence of IGH/CCND1 fusion. The presence of an unbalanced or balanced t(11;14) would be indicated by 1 or 2 red/green (yellow) fusion signals, respectively. (B) Representative interphase cell demonstrating 2 intact yellow fusion signals (5′ CCND1 probe, ∼384 kb [green]; 3′ CCND1 probe, ∼256 kb [red]) using a CCND1 break-apart probe (BAP) set. The presence of a CCND1 rearrangement would be indicated by separated or single green (5′ CCND1 probe) and red (3′ CCND1 probe) signals. (C) Junction plot demonstrating the insertion of an intact CCND1 gene into the IGH locus. In addition, loss of a portion of distal IGH-J, IGH-D, and a portion of the proximal IGH-V region was detected (2N indicated by the dashed gray horizontal line below the IGH locus label). (D) A depiction of the cryptic CCND1 insertional event. Dashed horizontal red lines on the normal chromosome 11 indicate breaks encompassing the CCND1 gene and insertion into the IGH locus located on the der(14). The black dots on IGH locus indicate enhancer elements. The IGH segment between the horizontal dashed red lines on chromosome 14 was deleted. The IGH/CCND1 D-FISH and CCND1 BAP footprints are indicated by the solid green/red and striped green/red horizontal bars, respectively. Insertion of the CCND1 gene into the IGH locus is unappreciable by FISH because of the minimal size of the inserted CCND1 gene segment. The secondary 12p/der(14q) translocation (not shown) also accounts for the extra IGH signal observed by IGH/CCND1 D-FISH studies.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/8/10.1182_bloodadvances.2019031450/2/m_advances031450f2.png?Expires=1768774387&Signature=QtYrFhMNTqdWZp8cL825GTFDRoxjQ6yG~gOIb3bexEmLkvpV40cicQg8Ju5zXJCqAPVS-sRSttd2DWEkbThoEZc~vedAN8851ujz5FQt8APi4bhoBBVPtsfde6yFE8JxBbWjuB~vdC8SxU~NhLQNN4E6W4So6NvlXb5ckSxlvR-xpuoZNM6wnBw~mj96kDEq9fFIHPN5k0o~PZ3E4qwUdZ3Y9lxu9F1~BNBrv1ZTZrDQwVCeccxFW3IxziSlKMZqs1mV4kZikIwC-hOkMv3KdhUyEfLZqTU7vJU8IH0ss72-KDf94IWCUiJergm7r2bVf5xQKG493YIXa7DJA8HnyA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)

