


Abstract
Sickle cell disease (SCD) is an autosomal-recessive hemolytic disorder with high morbidity and mortality. The pathophysiology of SCD is characterized by the polymerization of deoxygenated intracellular sickle hemoglobin, which causes the sickling of erythrocytes. The recent development of metabolomics, the newest member of the “omics” family, has provided a powerful new research strategy to accurately measure functional phenotypes that are the net result of genomic, transcriptomic, and proteomic changes. Metabolomics changes respond faster to external stimuli than any other “ome” and are especially appropriate for surveilling the metabolic profile of erythrocytes. In this review, we summarize recent pioneering research that exploited cutting-edge metabolomics and state-of-the-art isotopically labeled nutrient flux analysis to monitor and trace intracellular metabolism in SCD mice and humans. Genetic, structural, biochemical, and molecular studies in mice and humans demonstrate unrecognized intracellular signaling pathways, including purinergic and sphingolipid signaling networks that promote hypoxic metabolic reprogramming by channeling glucose metabolism to glycolysis via the pentose phosphate pathway. In turn, this hypoxic metabolic reprogramming induces 2,3-bisphosphoglycerate production, deoxygenation of sickle hemoglobin, polymerization, and sickling. Additionally, we review the detrimental role of an impaired Lands’ cycle, which contributes to sickling, inflammation, and disease progression. Thus, metabolomic profiling allows us to identify the pathological role of adenosine signaling and S1P-mediated erythrocyte hypoxic metabolic reprogramming and hypoxia-induced impaired Lands' cycle in SCD. These findings further reveal that the inhibition of adenosine and S1P signaling cascade and the restoration of an imbalanced Lands' cycle have potent preclinical efficacy in counteracting sickling, inflammation, and disease progression.
Introduction
In the 20th century, the focus on sickle cell disease (SCD) was a milestone in the history of medical science. Using a light microscope, James B. Herrick identified “sickle-shaped” erythrocytes isolated from a patient with respiratory symptoms.1 Further studies were performed, which determined abnormalities in β-hemoglobin (Hb) isolated from SCD erythrocytes.2,3 Moreover, there are findings that confirm that the differences in sickle Hb compared to normal Hb are due to ionizable amino acids.4,5 Further studies performed by Vernon Ingram, using innovative protein sequencing, identified an amino acid substitution of valine for glutamine at the sixth position on the β-globin chain (β6 glutamate → valine).6 Further studies performed by Max Perutz led to the revolutionary discoveries of the crystal structure of Hb, which provided a molecular insight into Hb allostery and sickle Hb polymerization7 . With these substantial contributions, SCD was identified as the first molecular disease.
As a result of the sickle mutation, deoxygenated HbS (deoxyHbS) forms long polymers, leading to the characteristic sickle-shaped erythrocytes and subsequent intravascular hemolysis. Without intervention, sickling-mediated hemolysis induces vaso-occlusion and leads to multiple organ damage that can cause severe pain and early death in patients.8 In the United States, ∼100 000 people living with SCD have an average life expectancy of ∼58 years.9,10 Because of the severe pathophysiological consequences of SCD, urgent emergency care is required to treat acute chest syndrome, infections, stroke, priapism, and chronic pain.8,11-13 Moreover, the incidence of SCD has increased significantly in recent decades, particularly in sub-Saharan Africa, the islands of the Caribbean, and the United States, leading to high health care costs that average >488 million dollars annually in the United States alone.9 It is predicted that, by the year 2050, nearly 10 million SCD patients will be needing treatment for serious life-altering complications.14 Although SCD was discovered >100 years ago, it is extremely disappointing that the precise treatment of SCD pain, end-organ damage, and other chronic conditions occurring in SCD is limited. In recent years, the use of cutting-edge metabolomics and state-of-the-art isotopically labeled nutrient flux analysis to monitor and trace intracellular metabolism in SCD mice and humans has led to multiple pioneering discoveries. These novel observations define the detrimental role of purinergic and sphingolipid signaling pathways that mediate hypoxic metabolic reprogramming, deoxyHbS polymerization, and sickling, the central pathogenic feature of SCD. These unexpected findings identify multiple innovative opportunities for disease management. Here, we review the metabolomics profiling in mice and humans with the SCD mutation and the functional role of metabolites in pathogenesis of the disease. In this review, we discuss the metabolic, molecular, and structural changes that underlie metabolic reprogramming. Preclinical and human translational studies focus on these newly identified signaling cascades and metabolic pathways as therapeutic targets in the medical management of SCD.
Metabolomics: the newest and most appropriate “omics” to accurately define the overall metabolite and functional changes in erythrocytes
Metabolomics is a robust and unbiased analytical method to accurately measure functional phenotypes that are the net result of genomic, transcriptomic, and proteomic changes, which responds faster to external stimuli than any other “ome.”15 This makes metabolomics particularly powerful for understanding the molecular basis underlying cellular and organ functional changes under physiological conditions in response to environmental stimuli, as well as in disease states.15-19 Metabolomics provides new molecular insights into the pathogenesis of disease and the physiological response to external stimuli, as well as identifies pathogenic and physiological biomarkers, for the diseases and physiological response. In turn, metabolomics profiling can reveal innovative therapeutic possibilities that can be useful in disease prevention and treatment by targeting an enzyme or pathway.15,20,21 Moreover, metabolomics profiling is especially appropriate for mature red blood cells (RBCs), in which gene-expression profiling is not an option because of the lack of a nucleus.
Metabolomics identifies overall metabolite changes in SCD mice
Unbiased high-throughput metabolomics has been used to define the blood metabolome of Berkeley SCD mice, an animal model for human SCD. These mice have transgenes containing the human α- and sickle β-Hb, which mimic the human SCD phenotype.22 Metabolomics revealed that 251 metabolites in 8 categories, including glycolysis, pentose phosphate pathway (PPP), amino acids, nucleotides, xenobiotics, lipids, fatty acids, and carbohydrates, are significantly altered in SCD mouse blood.23 Several metabolites identified in the screen include nucleosides (eg, adenosine), lipids (eg, sphingosine-1-phosphate [S1P], lysophospholipids, and free acyl fatty acids), and glycolytic intermediates (eg, 2,3-bisphosphoglycerate [2,3-BPG]), which are significantly elevated in SCD blood.23-25 In contrast, glutathione levels are significantly reduced.26 Thus, metabolomics profiling reveals overall specific metabolic changes in SCD mice.
Metabolomics analysis provides relevant clinical insight for predicting SCD pathophysiology in humans
Human metabolomics studies are applicable for assessing disease prognosis at the cellular level. In fact, changes in normal and sickle erythrocytes observed in patients reveal distinct changes in metabolic pathways that contribute to the disease in mice.23,26,27 Metabolomics analysis reveals elevated levels of endogenous glycolytic intermediates, along with reduced PPP metabolites, in human sickle erythrocytes compared with human nonsickle erythrocytes.26 Consistent with enhanced glycolysis, elevated levels of 2,3-BPG were identified in SCD patients, which confirms our mouse findings.26 Reduced levels of antioxidative metabolites, such as glutathione, were identified in the human metabolomics analysis, consistent with reduced flux through the PPP.26 These changes (ie, increased 2,3-BPG and reduced glutathione) are distinctly related to SCD progression. Additionally, elevated levels of carnitines and choline, which are required for membrane turnover and increased expression of metabolic precursors involved in polyamine synthesis, were identified in SCD erythrocytes.26 Overall, metabolic profiling in SCD patients provides a valuable assessment of biomarkers that contribute to SCD pathophysiology. In the following sections, we review the pathogenic nature, which reveals the molecular and metabolic alterations that occur in SCD mice and humans.
Excess extracellular adenosine is detrimental in SCD via ADORA2B activation
Acutely accumulated adenosine is a beneficial response to hypoxia or tissue injury by promoting vasodilation, increased oxygen delivery to tissues, and wound healing.28-30 However, persistently elevated adenosine can be detrimental and contribute to multiple disease conditions, such pulmonary fibrosis, chronic kidney disease, and preeclampsia.31-35 Adenosine signals via 4 G protein–coupled receptors, ADORA1, ADORA2A, ADORA2B, and ADORA3, which are expressed in a variety of cell types.36 Among the 4 adenosine receptors, ADORA2B has the lowest affinity for adenosine and is activated by substantial accumulation of extracellular adenosine. In the next section, we focus on the detrimental role of excess adenosine signaling in SCD.
Metabolomics profiling allows us to identify that excess adenosine contributes to sickling via ADORA2B signaling
Unbiased metabolomics profiling of whole blood showed that adenosine is increased in SCD mice compared with controls. Follow-up studies revealed that sustained increases in plasma adenosine activated signaling pathways in erythrocytes that contribute to sickling.25 Fortunately, targeting excess adenosine with a US Food and Drug Administration (FDA)–approved drug, polyethylene glycol–modified adenosine deaminase (PEG-ADA), lowers adenosine levels in SCD mice.25,37 Treating SCD mice with PEG-ADA enzyme therapy resulted in a reduction of sickling and subsequently attenuated pathological features associated with SCD.25
Using 4 adenosine receptor–deficient mice, follow-up studies revealed that elevated adenosine signaling via specific ADORA2B receptor promotes the production of 2,3-BPG, an erythroid-specific glycolytic metabolite, which negatively modulates Hb–O2 binding affinity.25 Preclinical studies showed that interfering with ADORA2B activation using a specific antagonist, PSB1115, decreased 2,3-BPG levels, deoxyHbS polymerization, and sickling, thus attenuating multiple tissue damage in SCD mice. Similarly, plasma adenosine is increased in patients with SCD, and inhibition of ADORA2B-mediated induction of 2,3-BPG protects against hypoxia-induced sickling in cultured human sickle erythrocytes. Altogether, these results indicate that an increase in adenosine signaling via ADORA2B-receptor activation mediates an elevation of 2,3-BPG levels in erythrocytes, which is detrimental in SCD, and that targeting this pathway may be an effective treatment for SCD.25
Ecto-nucleosidase CD73 ablation ameliorates SCD pathological phenotypes
Adenosine is a critical signaling molecule that plays multiple roles in orchestrating a cellular response in tissues under stress.30 Under these conditions, damaged cells release adenosine triphosphate, which is subsequently metabolized to adenosine diphosphate and adenosine monophosphate (AMP) by the ecto-nucleosidase CD39. Ecto-nucleosidase CD73 then converts AMP into adenosine, where extracellular adenosine activates 1 of its 4 G protein–coupled receptors.38,39 It was demonstrated that soluble CD73 activity is increased and correlates significantly with adenosine plasma concentration in Berkeley SCD-transgenic mice and SCD patients.40 Genetic and pharmacologic studies in SCD mice revealed that deletion of CD73 and inhibition of CD73 activity reduced erythrocyte sickling and hemolysis and improved the life span of erythrocytes.40 Additionally, CD73 deficiency and the inhibition of CD73 by adenosine 5′-(α,β-methylene) diphosphate (a CD73-specific inhibitor) in SCD mice led to the reduction of multiple organ damage, particularly in the lungs.40 Also, reduced vascular leakage and ameliorated tissue inflammation in other organs were observed.40 Mechanistically, mouse and human in vitro studies revealed that ADORA2B activation induces AMP-activated protein kinase (AMPK) and BPG mutase activation, which results in an increase in the production of 2,3-BPG and promotes the release of oxygen from oxygenated HbS.40 The resulting deoxyHbS molecules form insoluble polymers that distort the erythrocytes and cause sickling.41 Overall, these findings revealed that elevated CD73 activity underlies increased plasma adenosine production and that increased adenosine signaling via ADORA2B contributes to sickling by inducing BPG mutase activity mediating 2,3-BPG production in an AMPK-dependent manner. These findings immediately offer multiple potential therapeutic targets in SCD (Figure 1).
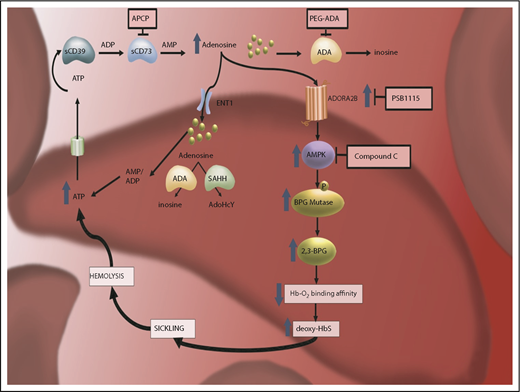
The discovery of pathogenic nature of elevated plasma adenosine signaling via ADORA2B receptor activation in sickling reveals multiple innovative therapeutic targets for SCD. CD73-dependent elevated extracellular adenosine signaling via ADORA2B receptor in RBCs leads to activation of AMPK, subsequently increased BPG mutase activity, elevated 2,3-BPG production, and, eventually, increased deoxyHbS polymerization and sickling. Thus, CD73–ADORA2B–AMPK signaling cascades are innovative therapeutic targets to counteract sickling. ATP, adenosine triphosphate.
The discovery of pathogenic nature of elevated plasma adenosine signaling via ADORA2B receptor activation in sickling reveals multiple innovative therapeutic targets for SCD. CD73-dependent elevated extracellular adenosine signaling via ADORA2B receptor in RBCs leads to activation of AMPK, subsequently increased BPG mutase activity, elevated 2,3-BPG production, and, eventually, increased deoxyHbS polymerization and sickling. Thus, CD73–ADORA2B–AMPK signaling cascades are innovative therapeutic targets to counteract sickling. ATP, adenosine triphosphate.
An unexpected observation of priapic features in ADA-deficient mice led to the discovery that excess adenosine signaling via ADORA2B promotes priapism and penile fibrosis in SCD mice
Priapism is define as prolonged undesired penile erection. More than 40% of male SCD mice exhibit this complication.42 No effective treatment is available for priapism because of the lack of understanding of the underlying mechanism that promotes priapism. Adenosine is a purine nucleoside that is irreversibly metabolized to inosine by adenosine deaminase (ADA). ADA deficiency in humans and mice results in chronically high adenosine levels that contribute to a variety of phenotypes via adenosine receptor activation.37 Notably, priapism was observed in ADA-deficient mice, which prompted the initial discovery that elevated adenosine signaling via the ADORA2B adenosine receptor mediates the transforming growth factor-β signaling cascade in the corpus cavernosum of male SCD mice, which promotes cyclic AMP and guanosine 3′,5′-cyclic monophosphate production and prolongs vasodilation in a protein kinase A (PKA)– and phosphodiesterase-dependent manner.43 Without interference, persistently accumulated plasma adenosine mediates ADORA2B receptor activation, which led to penile fibrosis and erectile dysfunction in ADA-deficient mice and SCD mice44 (Figure 2).
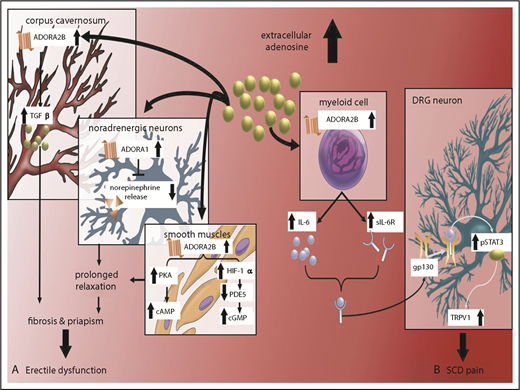
Excess extracellular adenosine signaling via ADORA2B receptor contributes to priapism, penile fibrosis, and pain in SCD mice. (A) Excess extracellular adenosine activates ADORA2B receptor in corpus cavernosum and smooth muscle relaxation, contributing to penile fibrosis and priapism, respectively, in SCD. (B) Excess extracellular adenosine activates ADORA2B receptor to induce secretion of IL-6 and sIL-6R from myeloid cells. IL-6 and sIL-6R form a complex that can transactivate gp130 in DRG neurons. gp130 activation leads to activation of phosphorylated STAT3 (pSTAT3), which, in turn, increases TRPV1 gene expression in DRG neurons and overall nociception in SCD mice. ADORA2B–IL-6–sIL-6–gp130 signaling networks are innovative therapeutic targets for priapism and chronic pain. cGMP, guanosine 3′,5′-cyclic monophosphate; PDE, phosphodiesterase.
Excess extracellular adenosine signaling via ADORA2B receptor contributes to priapism, penile fibrosis, and pain in SCD mice. (A) Excess extracellular adenosine activates ADORA2B receptor in corpus cavernosum and smooth muscle relaxation, contributing to penile fibrosis and priapism, respectively, in SCD. (B) Excess extracellular adenosine activates ADORA2B receptor to induce secretion of IL-6 and sIL-6R from myeloid cells. IL-6 and sIL-6R form a complex that can transactivate gp130 in DRG neurons. gp130 activation leads to activation of phosphorylated STAT3 (pSTAT3), which, in turn, increases TRPV1 gene expression in DRG neurons and overall nociception in SCD mice. ADORA2B–IL-6–sIL-6–gp130 signaling networks are innovative therapeutic targets for priapism and chronic pain. cGMP, guanosine 3′,5′-cyclic monophosphate; PDE, phosphodiesterase.
Elevated extracellular adenosine contributes to SCD pain via ADORA2B signaling
SCD pain is a chronic condition that results from increased nociceptor firing, increased responses to pain-evoking stimuli, and decreased thresholds for mechanical and thermal stimuli. Increased nociceptor sensitization mediated by painful stimuli contributes to pain responses in SCD. Particularly, transient receptor potential vanilloid 1 (TRPV1), a calcium-mobilization channel, induces nociceptive sensitization in the dorsal root ganglion (DRG) neurons, which contributes to SCD pain.11 It has been reported that increased TRPV1 responses in DRG neurons contribute to pain in SCD and ADA-deficient mice.45 Decreasing adenosine with PEG-ADA enzyme therapy or interfering with ADORA2B activation in SCD and ADA-deficient mice resulted in reduced TRPV1 responses in DRG neurons, decreased IL-6 levels, and lowered mechanical and thermal hypersensitivity.45 Although ADORA2B receptor expression is low in DRG neurons,45 excess adenosine signaling via ADORA2B receptors on myeloid cells promotes the secretion of soluble interleukin-6 receptor (sIL-6R) and interleukin-6 (IL-6), which form the sIL-6R–IL-6 complex to trans-activate gp130 and induce TRPV1 response in DRG neurons that promotes pain response45 (Figure 2). However, pharmacologic antagonism of ADORA2B receptor signaling by PSB1115 reduced pain sensitivity in ADA-deficient mice and SCD mice.45 Taken together, the sustained increase in adenosine signaling via ADORA2B contributes to sickling and multiple complications, including priapism, chronic pain, and tissue damage.
Metabolomics identifies that elevated S1P plays a pathological role in SCD pathophysiology
S1P is a bioactive sphingolipid that has multiple physiological and pathological roles.46-48 Metabolomics profiling has substantially advanced our understanding of its important role in SCD pathogenesis. In addition to adenosine, metabolomics revealed an elevated level of circulating S1P, a bioactive and abundant lipid, in SCD mice and humans.23 S1P is generated by sphingosine kinase-1 (SphK1) and SphK2. Only SphK1 is present in erythrocytes49 and, unlike other cell types, erythrocytes lack S1P-degrading enzymes.50 Thus, S1P is stored in erythrocytes at a relative higher concentration compared with other cell types.50 Additionally, the erythrocyte is considered the major cell type that maintains plasma S1P levels via export by a specific transporter, the major facilitator superfamily transporter 2b.51
Erythrocyte ADORA2B receptor–mediated activation of SphK1 underlies increased production of S1P in SCD
Human and mouse studies revealed that elevated plasma adenosine signaling via ADORA2B promotes the activation of SphK1 in normal and sickle erythrocytes.52 Subsequent studies revealed that PKA and extracellular signal-regulated kinase are essential intracellular signaling cascades that underlie ADORA2B-mediated activation of SphK1 in sickle and normal erythrocytes.52 These studies revealed that purinergic signaling components cross talk with the S1P signaling network via ADORA2B-dependent activation of SphK1 in erythrocytes (Figure 3).
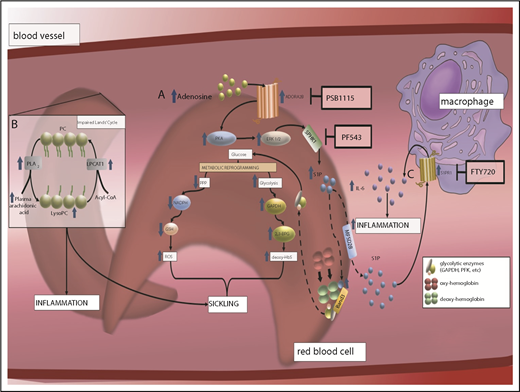
The discovery of an elevation of S1P and an impaired Lands' cycle in erythrocytes promoting sickling reveals multiple potential therapeutics to treat SCD. (A) Elevated plasma adenosine signaling via ADORA2B receptor underlies activation of SphK1 and increased S1P production in sickle RBCs in a PKA- and extracellular signal-regulated kinase (ERK)–dependent manner. Elevated intracellular S1P directly binds to deoxyHbS, which promotes deoxyHbS anchoring to membrane-bound band 3. This enhances the release of membrane-bound glycolytic enzymes to the cytosol, inducing metabolic reprogramming by accelerating glucose metabolism toward glycolysis instead of PPP, which induces 2,3-BPG production and nicotinamide adenine dinucleotide phosphate–mediated glutathione synthesis reduction. As such, elevated SphK1 mediates intracellular S1P production in sickle cells, which results in an increase of 2,3-BPG production and reactive oxygen species (ROS) generation. (B) Elevated erythrocyte membrane lysophosphatidylcholine (LysoPC) content and circulating erythrocyte arachidonic acid in sickle RBCs is the result of an impaired Lands’ cycle. Correcting an imbalanced Lands’ cycle by interfering with the activity of cytosolic PLA2 or inducing activation of lysophosphatidycholine acyltransferase 1 (LPCAT1), 2 key enzymes of the Lands’ cycle, led to a reduction of elevated RBC membrane LysoPC content and circulating arachidonic acid levels, which attenuated sickling. (C) Elevated plasma S1P signaling via S1PR1 contributes to a sustained inflammatory response and disease progression in SCD by reciprocal upregulation of IL-6 and S1PR1 expression in macrophages.
The discovery of an elevation of S1P and an impaired Lands' cycle in erythrocytes promoting sickling reveals multiple potential therapeutics to treat SCD. (A) Elevated plasma adenosine signaling via ADORA2B receptor underlies activation of SphK1 and increased S1P production in sickle RBCs in a PKA- and extracellular signal-regulated kinase (ERK)–dependent manner. Elevated intracellular S1P directly binds to deoxyHbS, which promotes deoxyHbS anchoring to membrane-bound band 3. This enhances the release of membrane-bound glycolytic enzymes to the cytosol, inducing metabolic reprogramming by accelerating glucose metabolism toward glycolysis instead of PPP, which induces 2,3-BPG production and nicotinamide adenine dinucleotide phosphate–mediated glutathione synthesis reduction. As such, elevated SphK1 mediates intracellular S1P production in sickle cells, which results in an increase of 2,3-BPG production and reactive oxygen species (ROS) generation. (B) Elevated erythrocyte membrane lysophosphatidylcholine (LysoPC) content and circulating erythrocyte arachidonic acid in sickle RBCs is the result of an impaired Lands’ cycle. Correcting an imbalanced Lands’ cycle by interfering with the activity of cytosolic PLA2 or inducing activation of lysophosphatidycholine acyltransferase 1 (LPCAT1), 2 key enzymes of the Lands’ cycle, led to a reduction of elevated RBC membrane LysoPC content and circulating arachidonic acid levels, which attenuated sickling. (C) Elevated plasma S1P signaling via S1PR1 contributes to a sustained inflammatory response and disease progression in SCD by reciprocal upregulation of IL-6 and S1PR1 expression in macrophages.
Elevated intracellular S1P induces sickling and inflammation in SCD by promoting metabolic reprogramming
Follow-up genetic and pharmacological studies revealed that genetic deletion of SphK1 or pharmacologic inhibition of SphK1 lowers S1P production, which has potent antisickling, antihemolysis, and anti-inflammation effects in SCD.23,53 Hypoxia mediates an increase in S1P, which promotes metabolic reprogramming in erythrocytes via band 3, the most abundant erythrocyte membrane protein, and releases glycolytic enzymes into the cytosol, thus promoting glycolysis.54,55 Isotopically glucose labeled flux analyses coupled with biochemical, cellular, and structural studies revealed that S1P binds to the central water cavity of deoxyHb, promotes the translocation of deoxyHb to the membrane, and triggers the release of glycolytic enzymes, such as glyceraldehyde-3-phosphate dehydrogenase, from membrane to cytosol. This process induces metabolic reprogramming, channeling glucose metabolism to glycolysis via the PPP. Overall, this metabolic shift contributes to RBC sickling due to an increase of intracellular S1P, which mediates 2,3-BPG production and reactive oxygen species generation53 (Figure 3).
Extracellular S1P signaling via its receptors contributes to increased inflammation and tissue damage that are independent of sickling in SCD
Similar to extracellular adenosine, extracellular S1P is a ligand for a family of S1P receptors (S1PRs). S1PR signaling has been identified to contribute to disease-related conditions, which include vasculature leakage, tissue injury, and pain.46-48 Among the 5 S1PRs, S1PR1 is ubiquitously expressed in multiple cell types and organs that have a high affinity for S1P.56,57 Although SphK1-mediated S1P production functions intracellularly to contribute to sickling by promoting metabolic switching of glucose metabolism toward glycolysis rather than the PPP, recent studies showed that elevated extracellular S1P signaling via S1PR1 in immune cells, particularly cells of the myeloid lineage, contributes to systemic inflammation and multiple tissue damage in SCD.58
Since the discovery of S1PRs, pharmacologic advances have been made to target individual S1PRs. FTY720 is an FDA-approved drug for the treatment of multiple sclerosis that was developed to target and inhibit S1PR1 signaling by internalization of S1PR1, followed by subsequent proteasome degradation.59 Although sickle erythrocytes have a much higher turnover than normal erythrocytes, it was determined that FTY720 did not reduce sickling.58 Instead, FTY720 was found to reduce systemic and local inflammation.58 S1P has been previously shown to activate receptors that induce sustained inflammation by persistently inducing IL-6 production in SCD mice.58 Genetic studies showed that genetic deletion of IL-6 reduced systemic inflammation, tissue damage, and kidney dysfunction in SCD mice. Moreover, sustained elevation of IL-6 levels was found to play a role in the increased S1PR1 gene expression in macrophages among several organs affected by SCD.58 Thus, increased S1P–S1PR1–IL-6 signaling functions as a malicious cycle that promotes chronic inflammation and tissue damage. Therefore, interruption of this malicious cycle by S1PR1 antagonism or IL-6 blockade is a potentially effective therapy for SCD.
Taken together, we showed that elevated SphK1 activity mediated by ADORA2B signaling underlies the increase of S1P in sickle erythrocytes. Elevated S1P functions intracellularly and contributes to sickling by promoting metabolic reprogramming. Increased plasma S1P functions via its receptor S1PR1 and contributes to systemic inflammation (Figure 3).
Impaired Lands’ cycle underlies sickling in humans and mice with SCD
The Lands’ cycle is essential to maintain the integrity of the plasma membrane. Cellular membranes contain glycerophospholipids, such as phosphatidylcholine, which contain 2 fatty acids linked to a glyceraldehyde-3-phosphate backbone at positions 1 and 2. Polyunsaturated fatty acids, including arachidonic acid, are usually located at position 2 of the glycerol phosphate backbone. De novo synthesis of glycerophospholipids is carried out by the Kennedy pathway, whereas removal and replacement of fatty acids at the sn-2 position are carried out by enzymes of the Lands’ cycle. The Lands’ cycle has been speculated to be involved in modifying the fatty acid composition of phospholipids, which function to regulate membrane homeostasis. Two enzymes responsible for regulating the Lands’ cycle are phospholipase A2 (PLA2) and lysophospholipid acyltransferases (LPLATs). LPAT1 functions as a repair enzyme that counteracts cytosolic PLA2–induced lysophospholipids to maintain the integrity of the plasma membrane.
Impaired Lands’ cycle contributes to sickling in SCD mice and cultured human SCD erythrocytes
Metabolomics revealed that erythrocyte membrane lysophosphatidylcholine and circulating erythrocyte-derived arachidonic acid levels are elevated in SCD erythrocytes, which suggests that the Lands’ cycle is impaired.24 In vivo mouse studies showed that elevated PLA2 contributes to sickling, inflammation, and multiple tissue damage in SCD mice.24 Cultured human sickle erythrocytes were treated with PLA2 small interfering RNAs, which reduced PLA2 activity and decreased sickling.24 Thus, this study reveals that an excessive accumulation of lysophospholipids in erythrocyte membranes is associated with an increase in PLA2. This study implies that inhibiting PLA2 with small interfering RNAs is a novel therapeutic approach for treating SCD (Figure 3).
Therapeutic potential and future directions
We highlighted several metabolic changes identified by metabolomics profiling in patients with SCD that appear to contribute to disease progression. A key metabolite is adenosine, a circulating purine nucleoside, which is posited to be pathogenic at chronically elevated levels by the activation of adenosine receptor signaling pathways in several tissues. A myriad of mouse genetic studies have shown that these pathologic pathways are CD73-ADORA2B–dependent signaling cascades that promote S1P-mediated sickling, pain, and inflammation via IL-6 production and AMPK activation in SCD.
A large number of pharmacological preclinical and human in vitro studies demonstrated that targeting CD73–ADORA2B and SphK1–S1P–S1PR1–IL-6 signaling cascades is a novel therapeutic approach for the treatment of SCD. For example, lowering adenosine levels to reduce ADORA2B-activated erythrocyte signaling pathways is a potential therapeutic option. A decrease in adenosine levels can be achieved by increasing its metabolic conversion to inosine or inhibiting its production. To achieve the former, adenosine can be decreased, but not totally eliminated, by an already clinically relevant drug used to treat ADA-deficient patients: PEG-ADA. In SCD, PEG-ADA reduced elevated adenosine levels and, in that way, reduced sickling, splenomegaly, multiple tissue damage, and pain.25 To lower adenosine levels by reducing adenosine production, one can consider using a CD73 inhibitor, such as adenosine 5′-(α,β-methylene) diphosphate, which has been shown to reduce sickling and multiple organ damage.
Alternatively, targeting ADORA2B receptors and their downstream targets, such as SphK1, has also been shown to ameliorate SCD phenotypes. ADORA2B antagonism by the PSB1115 compound also reduces sickling, inflammation, and pain in SCD mice. Elevated adenosine-ADORA2B signaling promotes sickling and inflammation, in part, through circulating S1P. S1P production by SphK1 can be targeted by PF543, a SphK1 inhibitor, which has been proven to have highly potent effects in decreasing inflammation. Sickle mice treated with PF543 showed dampened systemic and local inflammation, attenuated multiple tissue damage, and increased survival rates.52 The FDA-approved drug FTY720, an S1PR1 inhibitor, is also expected to be a promising alternative to suppress S1P signaling. With the discoveries made by metabolomics profiling, we are closer to understanding how multiple sickling-relating factors contribute to the pathogenesis of SCD, which sets up a stronger foundation to provide better treatment options for those suffering from SCD.
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (DK083559) and National Heart, Lung, and Blood Institute (HL114457, HL113574, HL137990, HL136969, HL137990-S, and HL136969-S) (Y.X.).
Authorship
Contribution: M.G.A. prepared the manuscript; J.M.M. prepared the figures and contributed to the writing of the manuscript; and Y.X. organized and revised the manuscript for final submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yang Xia, The University of Texas McGovern Medical School, 6431 Fannin, MSB 6.202, Houston, TX 77030; e-mail: yang.xia@uth.tmc.edu.

