Abstract
The clinical heterogeneity of the myelodysplastic syndromes (MDSs) relates to the recently discerned panoply of molecular abnormalities extant within this disease spectrum. Despite increasing recognition of these biologic abnormalities, very limited therapeutic options exist to exploit our increasing understanding of the molecular pathophysiology of MDS, with only 1 therapy (lenalidomide) particularly focused on a specific clinical patient subset (del(5q) cytogenetics) and 2 epigenetic modulators (azacitidine and decitabine) having been approved for treating these patients. This article will review the mutational and biologic landscape of these disorders, as well as the targeted therapeutics currently in clinical trials that are focused on attacking these features. Given the molecular complexity of these disorders and the limited repertoire of effective therapeutic agents, we will also discuss novel approaches attempting to determine potentially effective and personalized treatment options through complementary chemosensitivity and computerized signaling network screening for these disparate MDS patient subsets. Translational use of such resources, combined with the rapidly evolving next-generation molecular technologies, should prove useful in effectuating improved and more selective options for therapy.
Introduction
The myelodysplastic syndromes (MDSs) are a heterogeneous spectrum of chronic myeloid hemopathies with associated symptomatic cytopenias and substantial potential for evolution to acute myeloid leukemia (AML).1,2 Clinical characterization has demonstrated these patients to be within diverse prognostic risk groups, necessitating differing treatment strategies.1 Only 3 drugs are approved by the US Food and Drug Administration (FDA) for treating this group of disorders, with none approved in the past decade. The hypomethylating agents (HMAs) azacitidine and decitabine have response rates ∼40% for higher-risk (HR) patients, and lenalidomide has a response rate of 60% to 70% for del(5q) patients and 20% to 30% for non-(del5q) patients.3-6 However, given the substantial proportion of MDS patients whose disease is unresponsive to treatment or relapses following initial response, clinical trials with new and effective drugs are urgently needed.
Much of the difficulty in treating these patients relates to their biologic heterogeneity, with the patients having a cohort of disparate cytogenetic and myeloid cell mutational profiles.7 During the past decade, major strides have occurred in the molecular characterization of MDSs.8-11 These studies have shown the type and incidence of somatic mutations contributing to dysfunctional signaling pathways in MDSs, as well as their association with disease prognosis and responsiveness to certain drugs. In addition, data are emerging demonstrating immune-related mechanisms that inhibit the body’s ability to delete the malignant clones in MDSs.12
Given this critical new knowledge, a large variety of biospecific agents targeting these pathogenetic mechanisms in MDS are currently being evaluated in clinical trials. Encouraging results from these investigational studies are emerging, demonstrating the potential for using such agents that focus on targeting specific disease mechanisms to treat this group of disorders. This article will review the current state of treatment options for MDS patients using these novel agents.
Molecular genetic landscape of MDSs
More than 45 recurrently mutated pathogenic somatic genes have been identified in MDSs that are involved in a variety of functional categories, including histone modification, DNA repair, epigenetic regulation, RNA splicing, transcription, chromatin remodeling, and kinase signaling networks13 (Figure 1).
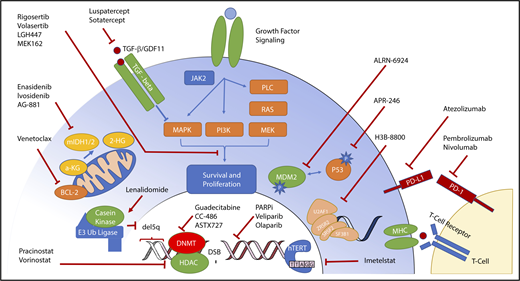
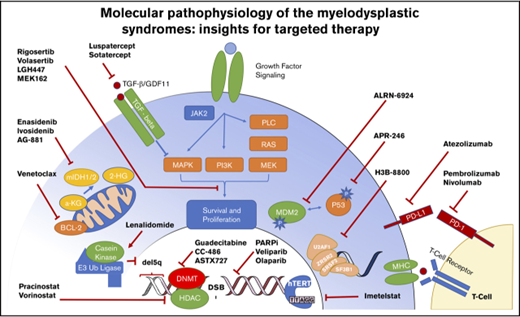
Molecular targets and therapeutics in MDS. The heterogeneity of molecular abnormalities extant within the MDS spectrum of disorders, involving a variety of somatically mutated genes and biologic functions, is demonstrated along with therapeutic agents in clinical trials focused on targeting these lesions.
Molecular targets and therapeutics in MDS. The heterogeneity of molecular abnormalities extant within the MDS spectrum of disorders, involving a variety of somatically mutated genes and biologic functions, is demonstrated along with therapeutic agents in clinical trials focused on targeting these lesions.
At the time of diagnosis, most cases of MDS are genomically complex, with many clones containing multiple cooperating mutations that can contribute to disease progression and/or relapse, although hematopoiesis is generally dominated by a specific clone.14 With the widespread adoption of targeted sequencing panels, it is now recognized that >80% of MDS patients harbor ≥1 known recurrently mutated gene.11 However, a given recurrent mutation is found in only a small proportion (∼10%) of the patients, for which ≥25 genes have prognostic implications. Frequently complementary mutations cooccur. However, certain mutations are noted to be largely mutually exclusive of one another (eg, the spliceosome genes).15 Single-cell sequencing studies of myeloid neoplasms are suggesting even more complex clonal architectures, as would have been predicted from bulk sequencing studies alone, with implications for disease monitoring and identification of treatment-resistant clones.16
The mutational dynamics of MDSs includes potential for change in clonal dominance with temporal evolution of the patients' specific mutations. Through serial monitoring of mutational status using next-generation sequencing (NGS), clonal evolution of MDSs can be followed over time. Administration of disease-altering therapy acts as a selective pressure to potentially skew the relative proportions of existing clones and leads to emergence of new clones associated with treatment resistance. In the following sections, we will review the evolving understanding of the recurrently mutated intracellular functional pathways that are frequently implicated in MDSs and novel therapies targeting these molecular defects. In addition, drugs capable of modifying potentially toxic marrow microenvironmental influences for erythropoiesis will be discussed (Table 1).
DNA maintenance and repair abnormalities
Maintaining the fidelity of the genome is critical for proper eukaryotic cell function. DNA damage can result from endogenous cellular processes or exogenous insults, such as cytotoxic chemotherapy and radiation. DNA double-strand breaks (DSBs) are 1 of the most threatening forms of genomic damage and are repaired by homologous recombination (HR) and nonhomologous end-joining (NHEJ).17 HR is the preferred cellular DNA repair process because it utilizes homologous DNA, typically from the sister chromatid, as a template for repair. Alternative pathways, such as NHEJ, repair DSBs in a crude fashion by joining broken DNA ends without effective error-suppression mechanisms. Any deficiency in HR leads to increased reliance on error-prone pathways like NHEJ and the introduction of mutagenic deletions and insertions.18
MDS pathogenesis is a complex process that involves multiple steps through a sequence leading to accumulation of genetic lesions in the DNA that alter cellular functions engendering emergence of premalignant clones. DSBs are the most severe type of DNA damage, which when not repaired can lead to chromosomal instability and emergence of chromosomal abnormalities. Investigations have implicated the HR and NHEJ mechanisms as being associated with the susceptibility, pathogenesis, and prognosis of MDSs.19,20 Recent data have emerged indicating that IDH1/2 mutations can lead to decreased HR repair and increased sensitivity of MDS cells to poly-ADP ribose polymerase (PARP) inhibitors and other DNA-damaging agents (temozolomide, cisplatin, daunorubicin).21
Defects in the HR pathway can be therapeutically exploited by overwhelming already defective DNA-repair mechanisms (platinum salts and alkylators) or by selectively increasing the reliance of tumor cells on alternative error-prone repair pathways.22,23 Early experience with the PARP inhibitor veliparib in combination with temozolomide showed modest signs of efficacy in a population of refractory or relapsed (R/R) AML or post-MDS patients, with complete remission (CR) rates of 17%. In this study, treatment-induced histone H2AX phosphorylation (a marker of DSBs) was associated with increased response. Most patients who achieved CR in this study had secondary or treatment-related AML in the setting of a prior diagnosis of MDS or chronic myelomonocytic leukemia (CMML).24 Additional studies of PARP inhibitors alone and in combination with HMAs in MDS are ongoing (Table 1).
Telomerase dysfunction
Defective maintenance of telomere integrity is a hallmark of cancer and is implicated in MDS pathogenesis. In MDSs, telomere erosion and dysfunction potentiate persistent DNA damage and accumulation of molecular alterations.25-27 Evidence suggests that telomere erosion can suppress hematopoietic stem cell self-renewal, repopulating capacity, and differentiation.28,29 Imetelstat is a first-in-class telomerase inhibitor that targets cells with short telomere lengths and highly active telomerase and has been shown in early clinical studies to have activity in myeloid malignancies.30,31 The role of imetelstat has been explored in MDS patients with low/intermediate-1 scores, based on the International Prognostic Scoring System, and R/R after erythroid-stimulating agents. Data from the first 32 patients in the open-label phase 2 portion of the phase 2/3 IMerge trial showed red blood cell transfusion–independence (TI) rates of 34% and erythroid hematologic improvement of 63% with imetelstat therapy at a median follow-up of 66 weeks.32 Reversible cytopenias were the most common adverse event. Extension of the IMerge trial is ongoing, randomizing patients to imetelstat vs placebo (NCT02598661).
TP53 mutation
Mutations in the TP53 tumor suppressor gene occur at different frequencies in a variety of MDS subsets. TP53 encodes a DNA-binding transcription factor that induces cell growth arrest, senescence, and cell death by apoptosis upon cellular stress, including oncogenic stress and DNA damage.33 In hematological malignancies, including MDS, TP53 mutations confer a poor prognosis and are particularly common in secondary MDS and a portion of patients with del(5q) cytogenetics.34 PRIMA-1 and the analog APR-246 (PRIMA-1MET) restore wild-type conformation of mutant p53 and showed activity in preclinical and early clinical studies, including a 60% response rate (partial response/CR) in p53-mutant ovarian cancer patients in combination with platinum-based chemotherapy.35,36 A phase 1/2a clinical trial with APR-246 in hematologic malignancies reported no major adverse effects, with a phase 2 trial in MDS now ongoing.37 Another approach to reactivate p53-mediated tumor suppression is to inhibit the frequently overexpressed p53 suppressor proteins MDMX and MDM2 in tumors. ALRN-6924, a cell-penetrating stapled α-helical peptide, disrupts the interaction between p53 and these endogenous inhibitors, thereby reactivating p53-mediated tumor suppression in AML cells.38 Phase 1/2 clinical testing with this drug is ongoing (Table 1).
Defective apoptosis
The antiapoptotic protein BCL-2 is overexpressed in hematologic malignancies, where it has been implicated in the maintenance and survival of myeloid cells, therapeutic resistance, and poor clinical outcomes. In a single-arm phase 2 study of the antiapoptotic agent venetoclax, single-agent activity was moderate in a population of R/R patients with refractory myeloid neoplasms (n = 32), including 12 (37.5%) patients with MDS. Of note, 33% of patients with isocitrate dehydrogenase 1/2 (IDH1/2) mutations achieved CR in this study, suggesting increased activity in this subgroup.39 Mutant IDH1/2 proteins alter the epigenetic landscape in AML cells through production of the oncometabolite (R)-2-hydroxyglutarate (2-HG), sensitizing these cells to Bcl-2 inhibition by venetoclax (ABT-199).40 This sensitization effect is induced by 2-HG–mediated inhibition of the activity of cytochrome c oxidase in the mitochondrial electron transport chain; suppression of cytochrome c oxidase activity lowered the mitochondrial threshold to trigger apoptosis upon BCL-2 inhibition. These findings are potentially valuable for considering the use of Bcl-2 inhibitors in IDH-mutated patients.
Marked synergistic activity has also been seen in in vitro and in vivo models of BCL-2 inhibition with lower-intensity therapies, such as low-dose cytarabine and HMAs. Phase 1b data have suggested a high degree of clinical activity of these combinations, especially in higher-grade myeloid malignancies, including MDS with excess blasts and AML, with 61% of patients achieving a CR or incomplete CR.41 The combination of venetoclax with HMAs or low-dose cytarabine has shown encouraging responses in R/R AML and MDS patients.42 These responses are predominantly evident in the upfront setting. Extensions of these studies are ongoing.
Epigenetic abnormalities
Aberrant differentiation in MDS is often related to abnormal DNA methylation and mutations in genes that regulate epigenetic programs (TET2 and DNMT3a, involved in DNA methylation control; EZH2 and ASXL1, involved in histone methylation control).43 This epigenetic nature of MDS may explain the finding that it is a disease most responsive to DNA methylation inhibitors (HMAs). Progression in MDS is characterized by acquisition of further epigenetic defects and mutations in growth-controlling genes that may alter the proliferation/apoptosis balance, thus contributing to the development of AML.
Azanucleosides, pyrimidine analogs of the nucleoside cytidine, are potent inhibitors of DNA methyl transferase and are referred to as HMAs. As indicated above, the azanucleosides now used clinically are 5-azacitidine and 5-aza-2′-deoxycytidine (decitabine). Parenteral formulations of azacitidine and decitabine remain the only FDA-approved HMA agents available for the treatment of MDS. Interest in oral formulations of HMA therapy has been increasing, for patient-related logistical reasons and based on preclinical data suggesting that extended dosing associated with increased drug exposure to leukemic cells was beneficial. Multiple such HMAs are in various stages of clinical development. Oral azacitidine (CC-486) has been evaluated in a phase 1/2 study in hematologic malignancies, showing good tolerability and overall response rates (ORRs) of 36% (14-day schedule) and 41% (21-day schedule) in lower-risk (LR) MDSs.44 CC-486 is being evaluated in a phase 3 randomized double-blind placebo-controlled LR MDS therapy trial (AZA-MDS-003).
Guadecitabine (SGI-110) is a small dinucleotide composed of decitabine and deoxyguanosine. Deoxyguanosine prevents deamination of decitabine, prolonging in vivo exposure of cells to the drug. Phase 2 data in HR MDS, CMML, and low blast count AML patients R/R after HMA therapy showed a modest salvage response rate.45 Another oral decitabine combination (ASTX727), includes the cytidine deaminase inhibitor cedazuridine plus decitabine. In a recent phase 1/2 trial, effective in vivo levels of the drug combination were achieved, with a good ORR.46 Although these new agents are not mechanistically novel, their oral potency provides potential logistic advantages for drug usage.
IDH1/2 dysregulation
Dysregulation of cellular metabolism has recently become an accepted “hallmark” of cancer.47 The IDH family of isozymes (IDH1, IDH2, IDH3) normally functions to convert isocitrate to α-ketoglutarate via oxidative decarboxylation.48 Mutations in IDH1 and IDH2 were initially described in gliomas, with elegant work showing that these mutations were always heterozygous, with resulting neomorphic activity causing conversion of α-ketoglutarate to the oncometabolite 2-HG.49,50 Elevated 2-HG levels alter epigenetic regulation and block cellular differentiation, thereby promoting tumor development.51
IDH1 and IDH2 mutations occur in ∼20% of de novo AML,52 with a lower frequency of 5% to 12% in MDS.53-55 As in AML, IDH1/2 mutations in MDS patients appear to be associated with older age, higher platelet counts, normal karyotype, and comutations in DNMT3A, ASXL1, and SRSF2.54 Additionally, IDH1 and IDH2 mutations appear to be mutually exclusive to each other and to TET2.54 Prognostic implications of IDH1/2 mutations in MDS remain controversial but appear to have an overall negative impact, especially in LR disease.53,55-57
A better understanding of the pathophysiology of IDH1/2 mutations in MDS and AML have led to development of clinical IDH1 and IDH2 inhibitors (Table 1). Enasidenib (AG-221) is a covalent inhibitor of IDH2 and was the first approved compound for treatment of R/R AML with IDH2 mutations, based on the AG221-C-001 study, an open-label single-arm multicenter study including 199 adults with a demonstrable IDH2 mutation. In this study, patients with R/R AML were treated with a daily oral dose of enasidenib. After a median follow-up of 6.6 months, 23% of patients experienced CR or incomplete CR lasting a median of 8.2 months. Of the 157 patients initially dependent on red blood cell transfusion, 34% achieved TI, with 76% of these patients maintaining prolonged TI status.58 Differentiation syndrome occurred in 14% of patients. A decrease in IDH2 variant allele frequency was not associated with CR, suggesting that enasidenib promoted differentiation and did not necessarily have cytotoxic effects. In a subset of 15 MDS patients included in the original phase 1 trials, ORR was 53%, although no mutational signature was predictive of response. Ongoing studies of enasidenib in MDS include a phase 2 trial examining the role of enasidenib alone or combined with azacitidine for an HMA R/R cohort in HMA-naive patients. Additional studies with the IDH1 inhibitor AG-120 (ivosidenib) have generated similar results, with phase 1 data in R/R AML showing a 42% ORR and 22% CRs, with a median response duration of 8.2 months.59 However, persistence of the IDH mutations occurred in ∼80% of those with CRs, suggesting differentiation as the primary mechanism of action. Importantly, no co-occurring mutations predicted clinical response or resistance to therapy. Similar studies are ongoing for HR MDS.
Deregulated histone deacetylases
Histone acetylation facilitates active gene transcription that is highly regulated by histone deacetylases (HDACs). HDAC inhibition can restore normal acetylation of deregulated histone proteins and transcription factors. Given the potential biologic synergy of HDAC inhibitors with HMAs, vorinostat, a small molecule inhibitor of class I and II HDAC enzymes, was combined with azacitidine in encouraging phase 1 and 2 studies for HR MDS. However, a large randomized phase 3 study evaluating the impact of azacitidine plus vorinostat vs azacitidine alone in HR MDS and CMML patients demonstrated that the ORR between these groups was similar, as were remission duration and overall survival (OS).60 Of interest, in a subgroup analysis, ORR was higher in patients with the epigenetically dysregulated DNMT3A mutations. Additional HDAC inhibitors have been developed to determine whether other classes of these agents could be beneficially applied. However, studies with entinostat61 and pracinostat62 have similarly shown no clinical benefit when combined with azacitidine compared with azacitidine alone. Despite early positive findings, the randomized trials indicated the potential challenges in trial design and potential toxicities for use of this drug combination, particularly because suboptimal combinations of overlapping, rather than sequential, HDAC/HMA, were utilized.63-65
Abnormal signal transduction
Abnormalities in signal-transduction pathways involved in the pathogenesis and progression of a wide variety of neoplasias have been demonstrated in MDSs, including those for the serine/threonine kinase AKT, which functions as a critical mediator of signaling downstream of phosphatidylinositol 3-kinase (PI3K),66,67 PKCa,68 the mammalian target of rapamycin,69 aurora kinases,70 cyclin D1,71 and casein kinase (CK1α).72
Given these findings, several kinase inhibitors have been used to treat MDS patients. The small molecule compound rigosertib binds to the Ras-binding domain of multiple kinases, including RAF, PI3K, and other effectors of Ras-related GTPases, and induces inhibition of the PI3K and polo-like kinase pathways.67,73-76 In a phase 3 trial that randomized 299 MDS patients to rigosertib vs best supportive care (BSC), although the median OS in the rigosertib group was not significantly longer than in the control group, in a preplanned exploratory analysis, patients with primary HMA failure treated with rigosertib had a trend toward longer median OS (8.6 vs 5.3 months).76 Additionally, in a post hoc analysis of patients with very high risk status based on the revised International Prognostic Scoring System, improved survival was noted in the rigosertib group compared with the BSC group (median OS, 7.6 vs 3.2 months). A randomized phase 3 trial of rigosertib vs BSC (NCT02562443) is underway in this HR population to attempt to confirm the potential role of this agent.
Another kinase abnormality implicated in these patients relates to the findings in the MDS del(5q) subtype. Although the mechanisms underlying the anemia in these patients remain elusive, haploinsufficiency and dependence of the erythroid cells on CK1α (a gene within the common deleted region of del(5q) MDS) appear to be of central importance. Lenalidomide induces ubiquitination of CK1α by the E3 ubiquitin ligase cereblon, resulting in CK1α degradation.72 Such degradation in the haploinsufficient del(5q) cells sensitized these cells to lenalidomide therapy, providing a basis for the therapeutic effects of the drug in these patients. The E3 ubiquitin ligase RNF41 is a principal target that is responsible for erythropoietin receptor stabilization. Data have indicated that lenalidomide also has E3 ubiquitin ligase inhibitory effects that inhibit RNF41 autoubiquitination and promote membrane accumulation of signaling-competent JAK2/EpoR complexes that augment erythropoietin responsiveness.77 An initial phase 2 trial showed particular (∼65%) responsiveness of MDS patients with the del(5q31) chromosomal abnormality to lenalidomide, demonstrating a major reduction in transfusion requirements and reversal of cytologic and cytogenetic abnormalities in this subset of patients.4 Reduced, although meaningful responses, were also seen in non-del(5q) patients in phase 2 and randomized phase 3 trials.5,78 The negative impact of TP53 mutations on responsiveness and outcome postlenalidomide is notable for ∼30% of these patients.79
Spliceosome dysregulation
Alternative splicing of messenger RNA (mRNA) precursors is a critical cellular process common to most eukaryotic genomes. The spliceosome is a large ribonuclear–protein complex located in the cell nucleus that is composed of 5 small nuclear ribonucleoproteins and >200 polypeptides.80 Sequencing studies have indicated that spliceosome mutations are very frequent in MDS, with upward of 45% to 60% of patients having somatic mutations affecting a component of the spliceosome.9,81 Mutations are typically heterozygous single nucleotide polymorphisms that localize to well-characterized hotspot regions of SF3B1, SRSF2, U2AF1, ZRSR2, PRPF40B, SF3A1, SF1, and U2AF65. When mutated, normal intronic splicing cannot occur efficiently, leading to intron retention and introduction of stop codons, rendering pre-mRNA susceptible to non-sense–mediated decay.82
Animal models have suggested a critical role for splicing mutations in the development of myeloid neoplasms. Work with the mouse model of inducible SRSF2 P95H and U2AF1 S34F mutations suggested that development of MDS is due, in part, to splicing alterations leading to ineffective hematopoiesis.9,83 Further investigations evaluating large unbiased screens of small molecules with antitumor properties uncovered a class of compounds capable of binding with high affinity to the 3′ splice site, destabilizing U2 small nuclear ribonucleoprotein/pre-mRNA complexes and preventing spliceosome assembly. Further refinement of these molecules caused their increased stability and activity leading to development of sudemycin, spliceostatin A, meayamycin, E7107, and, most recently, H3B-8800.
In SRSF2 p95H–transgenic mice, E7107 resulted in reduced disease burden.84 Based on these results, E7107 entered phase 1 studies in patients with advanced solid tumors. Single-agent antitumor activity was observed, and corollary studies showed pre-mRNA splicing modulation in peripheral blood mononuclear cells, suggesting on-target activity. However, the unanticipated side effect of visual loss in 2 participants secondary to optic neuritis necessitated discontinuation of the trial.85 Preclinical models indicated that splicing modulators are selectively lethal to tumor cells via the mechanism of synthetic lethality.86 The only splice gene modulator compound in active clinical trials is H3B-8800, an orally available selective small molecule SF3B1 modulator. The compound is being evaluated in LR and HR MDS, CMML, and AML in a phase 1/2 open-label multicenter study (Table 1). Cohorts with canonical splicing mutations and wild-type patients are being evaluated for safety. Should safety data be favorable, the trial will continue to an expansion phase that will enroll cohorts enriched for patients with splicing mutations.
Inhibitory cytokine effects
Increased levels of the transforming growth factor β superfamily inhibitors of erythropoiesis (predominantly growth and differentiation factor-11) occur within MDS erythroid cells. Luspatercept (ACE-536) is a novel fusion protein that acts as a ligand trap and binds to and blocks this inhibitory factor’s Smad2/3 signaling, thus increasing erythropoiesis.87 Encouraging phase 2 data, with good drug tolerance, have been shown in LR MDS patients who are nonresponsive to erythroid-stimulating agents, predominantly those with ring sideroblasts. These patients demonstrated an ∼60% erythroid response rate compared with ∼25% for those lacking this feature.88 A phase 3 randomized trial with luspatercept is currently completing analysis (NCT02631070). Similar phase 2 results, although with somewhat increased toxicity, have been reported for the chemically related compound sotatercept in the treatment of LR MDS.89
Immune dysregulation
Immunologic tolerance to cancer has major implications for the ability of tumors to survive, despite a variety of therapeutic approaches. A critical mechanism underlying this microenvironmental dysfunction relates to the ability of tumor cells to block immune checkpoints through expression of specific proteins that interfere with immune cell effector function. Expression of the lymphoid transmembrane PD-1 after T-cell activation is a normal physiological checkpoint that the immune system utilizes to avoid self-reactive T cells.90 However, tumor cells, through their generation and overexpression of ligands for PD-1 (eg, PD-L1 and PD-L2), exploit this pathway directly through expression of PD-L1/PD-L2 or indirectly by recruiting regulatory T cells that express PD-L1, causing immune dysfunction.91-93 Recent clinical trials have used a number of checkpoint inhibitors to treat solid and lymphoid tumors with variable degrees of efficacy.91
Such approaches have recently been applied to myeloid malignancies. Phase 1 trials for treating MDS have been reported with several checkpoint inhibitors as targets, either alone or in combination with azacitidine. The combination has been used because HMAs increase the levels of checkpoint inhibitors.12,94 The agents, predominantly used in HMA-resistant disease, were pembrolizumab (PD-1 inhibitor), nivolumab (PD-1 inhibitor), and ipilimumab (CTLA-4 inhibitor). Pembrolizumab alone was used mainly for LR patients, whereas nivolumab and ipilimumab were used alone or with azacitidine for HR patients.95 These trials demonstrated generally low hematologic responses but tolerable safety profiles.44,96 A phase 1b/2 randomized trial of atezolizumab, alone or with azacitidine, for HMA R/R or naive MDS patients is ongoing (NCT02508870).
Personalized therapy selection in MDS
As discussed in this review, in recent years the previously homogeneous clinical view of MDS has been supplanted by heterogeneous groupings defined by disease-related cytogenetic and molecular alterations. Despite our increasing molecular understanding of disease, response rates remain suboptimal (CR < 20%), even in molecularly predefined subgroups of patients treated with some of the novel agents. Beyond the compounds discussed above, multiple drugs targeting various dysregulated pathways within MDS patients have FDA approvals for other indications. However, despite advances in solid tumor and AML trial design, matching patients by genetic signature to targeted therapies (so-called “basket and umbrella trials”), such strategies have not had widespread adoption in MDS. Recently, 2 complementary strategies have emerged for potentially improving patient stratification in myeloid malignancies: ex vivo drug response profiling and in silico predictive simulation modeling. Increasing data suggest that such approaches may be useful in clinical personalized therapy selection for MDS patients.
Overview of in silico testing
In silico predictive simulation is a rapidly emerging technique for the modeling of a digital library of molecular targeted drugs on a cell signaling model informed by an individual tumor’s molecular and cytogenetic abnormalities. Using computer-simulated approaches, these techniques determine whether the patient’s gene mutations result in activated or inactivated proteins and then whether the protein is overexpressed or underexpressed, by utilizing information derived from the patient’s cytogenetics and/or chromosome copy number variation data. Protein network maps of each patient’s mutatome depict the interactive nature of all predicted aberrant protein signaling pathways.97
Recently reported data have shown the ability of this technology to predict treatment response in MDS patients receiving azacitidine, decitabine, venetoclax, and lenalidomide.97-99 In 1 cohort of 46 del(5q) MDS patients, the in silico approach predicted responses to lenalidomide treatment in 80% of patients.97 These studies have demonstrated that a computational biology technology is able to detail the complexity of the MDS signaling networks to simulate and predict drug response. Further studies are ongoing utilizing such approaches as a clinical decision support tool.
Overview of ex vivo testing
Ex vivo chemosensitivity assays have been used in the study of myeloid neoplasms for >30 years. These assays use incubation of viable patient-derived mononuclear myeloid cells with various chemotherapeutic compounds, with efficacy of response measured by a variety of direct or indirect measures of cell viability, cell cycle arrest, or apoptosis. However, clinical implementation of these assays has been limited by lack of standardization, difficulty with automation, operator-dependent biases, difficulty maintaining living cells in culture, and slow turnaround time. Newer fully automated techniques, utilizing flow cytometric readouts, have begun to overcome many prior limitations.98,100,102
Preliminary correlative studies have suggested the ability of such testing to predict clinical response to chemotherapeutic agents. Techniques have been applied to primary MDS patient samples, with resulting ex vivo results identifying novel subgroups of MDS with unique drug-sensitivity patterns correlating with clinical responses.103 Additionally, this type of approach has recently been prospectively studied in patients with refractory AML using an ex vivo drug-sensitivity testing (DST) platform. The study showed treatment response and survival benefit of DST-guided therapy compared with patients treated according to non-DST recommendations.104
Authorship
Contribution: A.A. and P.L.G. designed and performed the research, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: P.L.G. receives research funds from H3 Biotech, Genentech, Notable Labs, and Celgene; and is a consultant to Novartis. A.A. is a consultant to Mission Bio Inc.
Correspondence: Peter L. Greenberg, Stanford University Cancer Center, 875 Blake Wilbur Dr, #2335, Stanford, CA 94305-5821; e-mail: peterg@stanford.edu.