

Abstract
Platelet-derived growth factor (PDGF)-B and PDGF β-receptor (PDGFRβ) deficiency in mice is embryonic lethal and results in cardiovascular, renal, placental, and hematologic disorders. The hematologic disorders are described, and a correlation with hepatic hypocellularity is demonstrated. To explore possible causes, the colony-forming activity of fetal liver cells in vitro was assessed, and hematopoietic chimeras were demonstrated by the transplantation of mutant fetal liver cells into lethally irradiated recipients. It was found that mutant colony formation is equivalent to that of wild-type controls. Hematopoietic chimeras reconstituted with PDGF-B−/−, PDGFRβ−/−, or wild-type fetal liver cells show complete engraftment (greater than 98%) with donor granulocytes, monocytes, B cells, and T cells and display none of the cardiovascular or hematologic abnormalities seen in mutants. In mouse embryos, PDGF-B is expressed by vascular endothelial cells and megakaryocytes. After birth, expression is seen in macrophages and neurons. This study demonstrates that hematopoietic PDGF-B or PDGFRβ expression is not required for hematopoiesis or integrity of the cardiovascular system. It is argued that metabolic stress arising from mutant defects in the placenta, heart, or blood vessels may lead to impaired liver growth and decreased production of blood cells. The chimera models in this study will serve as valuable tools to test the role of PDGF in inflammatory and immune responses.
Introduction
Platelet-derived growth factor (PDGF) constitutes a family of mitogens, primarily for mesenchymal cells, that includes at least 4 dimeric forms: PDGF-AA, -AB, -BB, and -CC, products of 3 distinct but highly related genes, respectively encoding the A-, B-, and C-chains.1-4 PDGF dimers activate 2 specific receptors,4-6 the PDGF α-receptor (PDGFRα), which binds the A-, B-, or C-chain, and the PDGF β-receptor (PDGFRβ), which binds only the B-chain. PDGF is a potent stimulant of mesenchymal cell proliferation, migration, and altered cell metabolism in vitro and in vivo.
Targeted disruption of PDGF-B or PDGFRβ genes results in highly similar or identical embryonic lethal mutant phenotypes, including microvessel leakage, hemorrhage, and microaneurysm formation. Disorders of the vascular system include lack of kidney glomerular mesangial cells,7-9 lack of microvascular pericytes in many tissues,10 hypoplasia or complete loss of vascular smooth muscle cells (vSMC) around small- and medium-sized arteries,11 dilated heart and aorta,7 and reduction of trophoblasts and pericytes in the placenta labyrinthine layer.12 In addition, each mutant displays anemia and thrombocytopenia.7 8
The hematopoietic defects may reflect a primary abnormality intrinsic to the hematopoietic system, or it may be secondary to cardiovascular or placental dysfunction. In principle, the hematopoietic defect in PDGF-B and PDGFRβ mutants may also contribute to the vascular dysfunction seen in these mice. The known expression patterns of PDGF-B and PDGFRβ are not helpful in discriminating between these possibilities because both vascular and hematopoietic expression are known to occur.
Circulating cells, especially monocytes and platelets, are a major source of PDGF after activation or injury.2 Cultured monocytes also express PDGFRβ,13 though the ability of monocytes to respond to PDGF has been controversial, and discrepant results may reflect the regulation of receptor expression.3 Natural killer cells and T cells also express PDGFRβ, and PDGF can modulate natural killer cell cytotoxicity14 and the pattern of T-cell lymphokines15 produced in vitro. PDGF is synthesized in megakaryocytes,16 and erythroid precursors can be induced to express PDGF after stimulation with erythropoietin in vivo.17,18 In mixed marrow cultures, PDGF stimulates primitive hematopoietic precursors,19 erythroid precursors,20,21 and megakaryocytopoiesis.22However, from existing data it is unclear whether PDGF directly affects hematopoietic function or indirectly stimulates hematopoietic precursors through the release of growth and differentiation factors from stromal cells.
In this paper, we address the nature and cause of the hematopoietic abnormalities in PDGF-B and PDGFRβ mutant embryos. Our results provide evidence against a critical role for hematopoietic PDGF-B or PDGFRβ in the hematopoiesis and maintenance of blood vessel integrity. The hematopoietic PDGF-B–deficient chimeras described in this study provide a potentially powerful tool for the study of macrophage-derived and platelet-derived PDGF-B in inflammation and wound healing.
Materials and methods
Mice
B6.SJL-Ptprca/Pep3b/Boyj(B6.Ly-5a) mice used as recipients were purchased from the Jackson Laboratory (Bar Harbor, ME) or were bred at the Fred Hutchinson Cancer Research Center (Seattle, WA). Mice were housed in groups of 5 under micro-isolated, pathogen-free conditions with twice-weekly cage changes and were given sterilized chow and acidified water (pH, 3.5) ad libitum.23 Hybrids (129SV × C57BL/6J) (Ly-5b) heterozygous for the PDGFRβ gene8were bred in the Department of Comparative Medicine, University of Washington (Seattle). Hybrids heterozygous for the PDGF-B gene7 were bred in the Department of Medical Biochemistry, University of Göteborg (Sweden). Recipients were transferred to conventional housing conditions 6 to 8 weeks after fetal liver cell or bone marrow transplantation.
Isolation of fetal liver cells and determination of PDGF-B and PDGFRβ genotypes
On day 16.5, embryos were isolated after PDGF-B+/− × PDGF-B+/− matings and after PDGFRβ+/− × PDGFRβ+/− matings. Embryos were delivered by cesarean section, euthanized, and subsequently sexed by abdominal dissection. PDGF-B−/− or PDGFRβ−/− mutants generally had smaller livers than wild-type (+/+) or heterozygous (+/−) littermates. Livers were disaggregated individually in sterile RPMI 1640 medium containing 20% fetal calf serum (Gibco BRL, Gaithersburg, MD). Liver cell suspensions from individual embryos were stored on ice for up to 3 days in a volume of 300 μL. Kidneys were inspected for the presence of hemorrhagic foci characteristic of PDGF-B−/− or PDGFRβ−/−mutants,7,8 and the phenotypes were recorded. All surgical procedures were performed under sterile conditions.
DNA was prepared from the tail of each embryo with a QIAamp tissue kit (Qiagen, Chatsworth, CA). The PDGF-B genotype was determined by polymerase chain reaction (PCR) using primers specific for the neomycin phosphotransferase cassette (5′-TGTTCTCCTCTTCCTCATCTCC-3′, 5′-ATTGTCTGTTGTGCCCAGTC-3′) and the wild-type exon 4-specific sequence (5′-AGCAGAGCCTGCTGTAATCGCCGAG-3′, 5′-TTGCACATTGCGGTTATTGCAGCAG-3′). Separate amplifications with each primer pair were performed for 25 to 30 cycles (96°C, 30 seconds; 61°C, 30 seconds; 72°C, 30 seconds), respectively, yielding 140-bp (neo) and 157-bp (exon 4) diagnostic amplification products. In the most recent transplantation experiments, genotyping of PDGF-B was performed using a polymerase chain reaction (PCR) mix containing one forward primer (5′-GGGTGGGACTTTGGTGTAGAGAGG-3′) and 2 reverse primers (5′-TTGAAGCGTGCAGAATGCC-3′, 5′-GGAACGGATTTTGGAGGTAGTGTC-3′), respectively, yielding 265-bp (wild-type–specific) and 624-bp (neo-specific) products (40 cycles: 96°C, 30 seconds; 59.5°C, 30 seconds; 64°C, 120 seconds).
The PDGFRβ genotype was assessed by PCR in separate amplifications using primer sets specific for knockout and wild-type alleles. A wild-type–specific primer (5′-GCACATTCTTGCCTGTCTGC-3′) in combination with a neo-specific primer 5′-TGGCTACCCGTGATATTGCT-3′) and PCR amplification (40 cycles: 94°C, 30 seconds; 60°C, 30 seconds; 72°C, 120 seconds) generated a 3-kb diagnostic product in mice carrying the PDGFRβ+/− or the PDGFRβ−/−genotype. An additional set of primers (5′-ATGGATGACACCTGGAGGCTGTAG-3′ and 5′-TTGCTGTCCGTGTTATGGCTCCTG-3′) was used to amplify the corresponding wild-type genomic sequence (37 cycles: 94°C, 60 seconds; 58°C, 50 seconds; 72°C, 120 seconds), also resulting in an approximately 3-kb product that was diagnostic for PDGFRβ+/+ or PDGFRβ+/− embryos.
Phenotypic appearance and genotyping results from each embryo were compared with specific attention to the presence of focal renal hemorrhage and the reduced liver size characteristic of null mutants. Only wild-type and null mutant fetal livers that showed concordant results were used for transplantation or stem cell assay. Concordance was high, and only the absence of an interpretable PCR result was grounds for excluding an embryo with a mutant phenotypic appearance. In the initial series of transplantation experiments, male and female fetal liver cells were transplanted into recipients of the same sex. In some later experiments, pools of male and female fetal liver cells were prepared.
In vitro colony-forming assays and erythropoietin enzyme-linked immunosorbent assay
Colony assays were performed as previously described for burst-forming unit-erythroid (BFU-E) and colony-forming units containing erythroid cells (CFU-E), granulocytes and macrophages (CFU-GM),24 and megakaryocytes (CFU-Meg).25Lysates of embryonic day (E)14.5 and E16.5 embryos were prepared and evaluated in a sensitive enzyme-linked immunosorbent assay for mouse erythropoietin26 using recombinant mouse erythropoietin as a standard.
Transmission electron microscopic analysis of fetal liver megakaryocytes
Half the E16.5 fetal livers were fixed overnight at 4°C with Karnovsky fixative, osmicated (2% OsO4 in veronal-acetate buffer, pH 7.4), stained in block with uranyl acetate, dehydrated in a graded series of ethanol, infiltrated with propylene oxide, and embedded in Epon. Thin sections were cut, stained with uranyl acetate and lead citrate, and examined with a JEOL transmission electron microscope (JEOL, Peabody, MA). At least 25 megakaryocytes were examined per fetal liver; in total, 6 fetal livers were examined (2 PDGFRβ−/−, 1 PDGF-B−/−, and 3 wild-type littermate controls).
Total body irradiation and fetal liver cell transplantation
Female and male Ly-5a recipients 8 to 12 weeks of age were prepared by total body irradiation in a single 14-Gy fraction from dual-opposed cobalt Co60 sources at an exposure rate of 20 cGy/min on the day before transplantation. Fetal cells were stained with propidium iodide (Sigma, St Louis, MO), and cell viability was analyzed by flow cytometry using a FACScan (Becton Dickinson, San Jose, CA). Aliquots containing 5 × 106 to 10 × 106viable fetal liver cells were injected into each recipient through the lateral tail vein.
Bone marrow transplantation
Bone marrow was flushed from the femurs23 of wild-type and PDGF-B−/− chimeras 20 weeks after fetal liver transplantation. Complete replacement of host with donor hematologic cells was confirmed in each donor before sacrifice. Marrow cells were washed in phosphate-buffered saline and injected into lethally irradiated (14 Gy) B6.Ly-5a recipients (10 × 106 viable cells/recipient).
Assessment of chimerism
The degree of chimerism after fetal liver cell or bone marrow transplantation was assessed at 28 days and at time points up to 107 days. Heparinized blood was obtained from the orbital venous plexus, and red blood cells were lysed in NH4Cl buffer. Leukocytes were stained with biotinylated Ly-5.1– and Ly-5.2–specific antibodies, as described previously.23 The Ly-5.1 antibody stained leukocytes derived from Ly-5a recipients, whereas the Ly-5.2 antibody stained leukocytes derived from Ly-5b donors. In some experiments, 2-color analysis was performed with fluorescein isothiocyanate –conjugated antibodies against CD3+ cells (hamster IgG mAb 145-2C11), B lymphocytes (rat B220, clone RA3-6B2; Pharmingen, San Diego, CA), CD4 (rat L3T4, clone H129.19; Pharmingen), and CD8 (rat Ly2, clone 53-6.7; Pharmingen). Stained cells were fixed in 1% paraformaldehyde and analyzed by flow cytometry. Scatter plots for monocytes, granulocytes, and lymphoid cells were determined by forward and side scatter characteristics, and the percentage of donor cells within each window was enumerated. Results were rounded to the nearest integer and were not corrected for background staining. For each experiment, thresholds for delineating positive and negative cells were determined by staining samples from appropriate positive and negative controls.23
Blood analyses
Venous blood (400 μL) from chimeras was obtained from the orbital venous plexus, and peripheral blood parameters were determined by using a Celldyn 3500 instrument (Abbott Diagnostics, Abbott Park, IL). Blood from decapitated E17.5 embryos was collected into microtiter EDTA tubes (Becton Dickinson, Bedford, MA) containing phosphate-buffered saline. Differential counting of nucleated cells was performed on May-Grünwald–stained blood films.
In situ hybridization
We applied a nonradioactive protocol for in situ hybridization using digoxigenin-labeled RNA probes (Boehringer Mannheim [now Roche Molecular Biochemicals], Indianapolis, IN) and for their detection on sections using alkaline phosphatase–conjugated antibodies.27 PDGF-B and PDGFRβ sense and antisense probes were generated as described previously.10 The results shown were obtained using 14-μm thick sections and interference contrast microscopy. As negative controls, the corresponding sense probes were used.
Results
Anemia, erythroblastosis, thrombocytopenia, and hepatic hypocellularity in PDGF-B−/− mutant embryos
Targeted deletion of the PDGF-B chain or the PDGFRβ was embryonic lethal, and null embryos displayed anemia and thrombocytopenia when evaluated at E18.7,8 To assess more completely the effect of full or partial disruption of the PDGF-B gene on the hematologic profile in embryos, we evaluated peripheral blood from decapitated E17.5 PDGF-B null mutant, heterozygous, and wild-type embryos (Figure 1). PDGF-B−/− mutant embryos showed a 50% reduction in the mean hemoglobin concentration (52 ± 11 g/L, n = 8,P < .0001) compared with that of wild-type controls (101 ± 7 g/L, n = 14), whereas heterozygous mutant embryos appeared mildly anemic (92 ± 11 g/L, n = 20,P = .0104 versus wild-type). Erythroblast counts ( × 109/L) were increased 15-fold in PDGF-B−/− mutants (21.8 ± 10.6, n = 7) compared with wild-type embryos (1.47 ± 1.54, n = 13, P < .0001) and 10-fold over heterozygous embryos (2.19 ± 1.62, n = 13,P < .0001). PDGF-B−/− mutants had low platelet counts (178.5 ± 69 × 109/L) compared with wild-type controls (345 ± 71 × 109/L,P < .0001). Heterozygous embryos showed a nonsignificant reduction in circulating platelets (331 ± 46 × 109/L,P = .587). The hematologic data in E17.5 PDGF-B−/− mutants are consistent with anemia, erythroblastosis, and thrombocytopenia.7 8

Anemia, erythroblastosis, and thrombocytopenia in PDGF-B null mutant embryos.
Peripheral blood hemoglobin concentration (left), platelet counts (center), and erythroblast (nucleated erythrocytes) counts (right) in wild-type(+/+), heterozygous(+/−), and PDGF-B mutant−/− embryos were measured at E17.5 in blood (*P < .001 by the Mann-Whitney test for comparison between wild-type and PDGF-B−/− mutants or between PDGF-B+/− and PDGF-B−/−; ‡P = .01 for comparison between PDGF-B+/+and PDGF-B+/−).
Anemia, erythroblastosis, and thrombocytopenia in PDGF-B null mutant embryos.
Peripheral blood hemoglobin concentration (left), platelet counts (center), and erythroblast (nucleated erythrocytes) counts (right) in wild-type(+/+), heterozygous(+/−), and PDGF-B mutant−/− embryos were measured at E17.5 in blood (*P < .001 by the Mann-Whitney test for comparison between wild-type and PDGF-B−/− mutants or between PDGF-B+/− and PDGF-B−/−; ‡P = .01 for comparison between PDGF-B+/+and PDGF-B+/−).
The fetal liver is the major site of hematopoiesis during midgestation in mice.28,29 We analyzed total cellularity and cell viability of mutant and wild-type livers after limited storage ex vivo (Table 1). Storage of individual fetal liver suspensions from E16.5 PDGF-B or PDGFRβ null mutants and their respective wild-type littermates for 24 to 48 hours was necessary for genotyping and transport between laboratories. Similar differences in the total number of viable fetal liver cells were observed in the absence of any storage time (data not shown). In all cases, a small proportion (less than 20%) of the viable nucleated cells was hepatocytes, as assessed by light microscopy. The total number of viable cells isolated from PDGF-B−/− livers was reduced by approximately 40% at E14.5 and E16.5 and by approximately 50% in PDGFRβ−/− livers at E16.5, compared with wild-type controls (Table 1). Because definitive embryonic erythropoiesis is dependent on erythropoietin,29 30 we also measured erythropoietin levels in E14.5 and E16.5 fetal livers. As shown in Figure 2, erythropoietin is increased in PDGF-B−/− embryos at both time points, as compared with PDGF-B+/+ embryos.
Reduced total cellularity of fetal livers from PDGF-B−/− or PDGFRβ−/− E16.5 embryos
| Genotype | Fetal liver cells (×106/liver) | |
|---|---|---|
| E14.5 | E16.5 | |
| PDGF-B+/+ | 39.5 ± 3.4 | 73.1 ± 3.2 |
| PDGF-B−/− | 24.7 ± 5.7* | 45.1 ± 3.9† |
| PDGFR-β+/+ | ND | 79.2 ± 2.1 |
| PDGFR-β−/− | ND | 39.8 ± 16.1‡ |
| Genotype | Fetal liver cells (×106/liver) | |
|---|---|---|
| E14.5 | E16.5 | |
| PDGF-B+/+ | 39.5 ± 3.4 | 73.1 ± 3.2 |
| PDGF-B−/− | 24.7 ± 5.7* | 45.1 ± 3.9† |
| PDGFR-β+/+ | ND | 79.2 ± 2.1 |
| PDGFR-β−/− | ND | 39.8 ± 16.1‡ |
Fetal liver cells were collected from E14.5 and E16.5 embryos and stored on ice for 24 to 48 hours. The percentage of viable E16.5 fetal liver cells obtained from PDGF-B+/+ embryos (n = 3) and PDGF-B−/− mutants (n = 5) after 48 hours of storage was 75.0% ± 1.6% and 75.6% ± 1.1%, respectively. Viability of PDGFRβ+/+ and PDGFRβ−/−fetal liver cells was 80.6% (pool of 4 livers) and 81.9% (pool of 4 livers), respectively.
P = .0003;
P< .0001;
P = .002; n = 6 for all groups.

Erythropoietin levels are increased in the fetal livers of E14.5 and E16.5 PDGF-B−/− embryos.
Livers from E14.5 and E16.5−/− and +/+embryos were snap frozen and kept at −80°C until analysis after lysis and homogenization in 150 mM NaCl, 66 mM EDTA, 10 mM Tris HCl (pH 7.4), 0.4% Na deoxycholate, 1% NP-40, and protease inhibitor cocktail (P-2714; Sigma). Erythropoietin activity was quantified using a combination of 2 commercially available erythropoietin enzyme-linked immunosorbent assay kits.26 Erythropoietin concentration was normalized to the total amount of protein in the sample using the Bio-Rad protein assay. The 2-tailed paired Student t test was used for statistical calculations (P = .023* andP = .030‡). Three animals of each genotype and age were tested. Error bars in the figure represent ±1 SD.
Erythropoietin levels are increased in the fetal livers of E14.5 and E16.5 PDGF-B−/− embryos.
Livers from E14.5 and E16.5−/− and +/+embryos were snap frozen and kept at −80°C until analysis after lysis and homogenization in 150 mM NaCl, 66 mM EDTA, 10 mM Tris HCl (pH 7.4), 0.4% Na deoxycholate, 1% NP-40, and protease inhibitor cocktail (P-2714; Sigma). Erythropoietin activity was quantified using a combination of 2 commercially available erythropoietin enzyme-linked immunosorbent assay kits.26 Erythropoietin concentration was normalized to the total amount of protein in the sample using the Bio-Rad protein assay. The 2-tailed paired Student t test was used for statistical calculations (P = .023* andP = .030‡). Three animals of each genotype and age were tested. Error bars in the figure represent ±1 SD.
Comparable hematopoietic progenitor colony formation with fetal liver cells from PDGF-B+/+ and PDGF-B−/− or PDGFRβ−/− embryos and normal megakaryocyte formation in PDGF-B and PDGFRβ null fetal livers
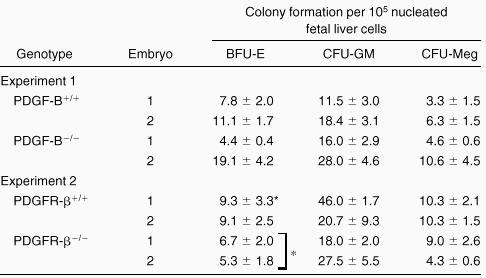
To investigate whether the fetal livers in PDGF-B−/−and PDGFRβ−/− embryos had been properly populated by hematopoietic progenitor cells, fetal liver cells were used for hematopoietic progenitor cell assays. As shown in Table2, we observed comparable hematopoietic progenitor colony formation for CFU-GM and CFU-Meg when equivalent numbers of fetal liver cells were assayed. A modest, but statistically significant (P = 0.04), reduction in BFU-E colony formation was seen with fetal liver cells from PDGFRβ−/− embryos (Table 2). No differences were observed in CFU-E in fetal liver cells from PDGF-B null (n = 3) and PDGFRβ null (n = 2) chimeras and wild-type (data not shown) littermate controls (n = 6). These results suggested that the expression of PDGF-B or PDGFRβ was not required for the development of these lineages.
Hematopoietic progenitor cell activity is not reduced in PDGF-B null or PDGFRβ null fetal livers
 |
|
Fetal liver cells from individual embryos (littermates) were suspended, and equivalent numbers of fetal liver cells were plated in methyl cellulose for determination of BFU-E, CFU-GM, and CFU-Meg. Colonies were scored from triplicate plates and are expressed ±SD. There is a modest reduction in BFU-E in PDGFRβ−/− fetal livers compared with PDGFRβ+/+ (*P= .04).
Because megakaryocytes are a major source of PDGF and platelets have been shown to express PDGFα receptors,31 we also examined megakaryocytes ultrastructurally from PDGF-B and PDGFRβ null fetal livers. We detected no defects by transmission electron microscopy in the formation of granules, demarcation membranes, or cytoplasmic fragmentation as compared with littermate wild-type control fetal livers (data not shown).
Generation of hematopoietic chimeras by transplantation of PDGF-B−/− or PDGFRβ−/− mutant fetal liver cells
To evaluate separately hematopoietic and vascular functions of PDGF-B and PDGFRβ, we constructed chimeras in which either PDGF-B or PDGFRβ was selectively disrupted in the hematopoietic compartment. The chimeras were prepared by transferring fetal liver cells into irradiated, H-2–compatible, wild-type recipients. In pilot dose-response experiments, the correlation between the administered irradiation exposure and the percentage donor cell reconstitution after 28 days was established for B6.Ly-5a recipients transplanted with wild-type B6.Ly-5b fetal liver cells. Complete (more than 98%) repopulation by donor granulocytes was achieved at an irradiation dose of 13 Gy or greater (data not shown). Therefore, we routinely administered a dose of 14 Gy in all transplantation experiments.
In 6 independent experiments, 5 × 106 to 10 × 106 viable fetal liver cells (E16.5, Ly-5b) from wild-type and PDGF-B−/−littermates (experiments 1-3) or from wild-type and PDGFRβ−/− littermates (experiments 4-6) were injected into 14-Gy–irradiated B6.Ly-5a recipients. Thirty-two recipients (experiment 1, n = 10; experiment 2, n = 10; experiment 3, n = 12) were engrafted with PDGF-B−/− fetal liver cells. Twenty recipients received PDGFRβ−/− fetal liver cells (experiment 4, n = 4; experiment 5, n = 8; experiment 6, n = 8), and 20 recipients were injected with control PDGF-B+/+ or PDGFRβ+/+ fetal liver cells obtained from littermate embryos. The survival rate beyond 14 days after transplantation was 90% to 100%. After the critical phase of acute irradiation sickness, all chimeras were healthy and appeared normal throughout the observation period of 4 to 12 months.
Kinetics of peripheral leukocyte reconstitution after transplantation of wild-type and PDGF-B−/− or PDGFRβ−/− fetal liver cells
Engraftment of fetal liver–derived granulocytes, monocytes, B cells, and T cells (CD4, CD8) was analyzed by 2-color staining of peripheral blood samples with an Ly-5.2-specific antibody and lineage-specific antibodies against B220 and CD3. By day 28 after transplantation, more than 98% of peripheral blood granulocytes, monocytes, and B cells were derived from the fetal livers of wild-type and PDGF-B−/− (Figure 3A) or wild-type and PDGFRβ−/− (Figure 3B) donors. Engraftment of donor-derived cells remained stable throughout observation periods up to 107 days, indicating that long-term reconstitution occurred. Results with wild-type and null mutant donors were indistinguishable.

Hematopoietic reconstitution after transplantation of PDGF-B−/− or PDGFRβ−/− fetal liver cells.
Fetal liver cells from wild-type (filled bars) or null mutant littermates (open bars) were collected from embryos (E16.5) from the (A) PDGF-B and (B) PDGFRβ breeding colony and transplanted into 14-Gy–irradiated B6.Ly-5a recipients (5 × 106 to 10 × 106/recipient). Data were obtained from 4 independent transplantation experiments. Each symbol represents the mean percentage from analysis of 4 to 10 recipients. PMN indicates polymorphonuclear cells; mono, monocytes.
Hematopoietic reconstitution after transplantation of PDGF-B−/− or PDGFRβ−/− fetal liver cells.
Fetal liver cells from wild-type (filled bars) or null mutant littermates (open bars) were collected from embryos (E16.5) from the (A) PDGF-B and (B) PDGFRβ breeding colony and transplanted into 14-Gy–irradiated B6.Ly-5a recipients (5 × 106 to 10 × 106/recipient). Data were obtained from 4 independent transplantation experiments. Each symbol represents the mean percentage from analysis of 4 to 10 recipients. PMN indicates polymorphonuclear cells; mono, monocytes.
Throughout the first 100 days after transplantation, the proportion of donor-derived T cells in the blood was lower than the proportion of donor-derived B cells and myeloid cells, but the proportion did not depend on donor genotype (Figure 3). The proportion of donor-derived T cells increased from 42.5% ± 6.9% (PDGF-B+/+ donors) and 39.7% ± 8.8% (PDGF-B−/− donors) at 28 days after transfer to 78.4% ± 3.8% and 71.9% ± 16.4%, respectively, by 45 days after transplantation (Figure 3A). Similar changes were observed in PDGFRβ chimeras (Figure 3B). By 100 days after transplantation, almost all T cells were donor-derived. At all time points, results with wild-type and null mutant donors were indistinguishable (Figure 3).
Normal peripheral blood parameters in PDGF-B−/− or PDGFRβ−/− mutant chimeras
All PDGF-B−/− and PDGFRβ−/−chimeras were healthy after they recovered from acute irradiation sickness, approximately 14 days after transplantation. Recipients transplanted from PDGF-B−/− and PDGFRβ−/−donors and their respective wild-type controls had identical appearances, weights, and behaviors at all time points after fetal liver transplantation. Bone marrow cross-sections from the chimeric mice 14-18 weeks after transplantation showed no overt phenotypic differences between the different chimeric combinations, which suggested that the hematopoietic activity in the bone marrow of null mutants was normal. No significant differences in red cell counts, platelet counts, or differential counts of granulocytes, lymphocytes, or monocytes were found between PDGF-B−/− and PDGF-B+/+ chimeras (Table 3) or PDGFRβ−/− and PDGFRβ+/+ chimeras (Table 4). These results indicated that normal hematopoietic functions of all major lineages were maintained in the absence of PDGF-B or PDGFRβ expression in hematopoietic stem cells.
Complete reconstitution of peripheral blood by E16.5 fetal liver cells from PDGF-B−/− embryos
| Blood parameter | Donor PDGF-B genotype | |||
|---|---|---|---|---|
| +/+ (n = 5) | −/− (n = 5) | |||
| Mean | ± SD | Mean | ± SD | |
| Hematocrit (%) | 40.50 | ± 1.40 | 42.00 | ± 1.50 |
| Erythrocytes (×1012/L) | 9.10 | ± 0.30 | 9.80 | ± 0.20 |
| Hemoglobin (g/dL) | 13.60 | ± 0.60 | 14.10 | ± 0.40 |
| MCV (fL) | 44.60 | ± 1.50 | 43.20 | ± 1.60 |
| MCHC (%) | 33.60 | ± 0.60 | 33.60 | ± 0.90 |
| WBC (×109/L) | 10.80 | ± 1.80 | 11.10 | ± 2.40 |
| PMN (×109/L) | 1.14 | ± 0.51 | 0.95 | ± 0.28 |
| Lymphocytes (×109/L) | 8.76 | ± 1.86 | 9.36 | ± 2.05 |
| Monocytes (×109/L) | 0.71 | ± 0.14 | 0.50 | ± 0.20 |
| Platelets (×109/L) | 542.00 | ± 67.00 | 670.00 | ± 137.00 |
| Blood parameter | Donor PDGF-B genotype | |||
|---|---|---|---|---|
| +/+ (n = 5) | −/− (n = 5) | |||
| Mean | ± SD | Mean | ± SD | |
| Hematocrit (%) | 40.50 | ± 1.40 | 42.00 | ± 1.50 |
| Erythrocytes (×1012/L) | 9.10 | ± 0.30 | 9.80 | ± 0.20 |
| Hemoglobin (g/dL) | 13.60 | ± 0.60 | 14.10 | ± 0.40 |
| MCV (fL) | 44.60 | ± 1.50 | 43.20 | ± 1.60 |
| MCHC (%) | 33.60 | ± 0.60 | 33.60 | ± 0.90 |
| WBC (×109/L) | 10.80 | ± 1.80 | 11.10 | ± 2.40 |
| PMN (×109/L) | 1.14 | ± 0.51 | 0.95 | ± 0.28 |
| Lymphocytes (×109/L) | 8.76 | ± 1.86 | 9.36 | ± 2.05 |
| Monocytes (×109/L) | 0.71 | ± 0.14 | 0.50 | ± 0.20 |
| Platelets (×109/L) | 542.00 | ± 67.00 | 670.00 | ± 137.00 |
Donor stem cells used for transplantation (10 × 106/recipient) were obtained from wild-type (+/+) or null mutant (−/−) littermates, and peripheral blood was evaluated 14 to 18 weeks after transplantation. MCV indicates mean corpuscular volume; MCHC, mean corpuscular hemoglobin concentration; WBC, white blood cells; PMN, polymorphonuclear leukocytes.
Fetal liver cells from PDGFRβ null embryos can completely repopulate the peripheral blood compartment of lethally irradiated hosts
| Blood parameter | Donor PDGFRβ genotype | |||
|---|---|---|---|---|
| +/+ (n = 5) | −/− (n = 5) | |||
| Mean | ± SD | Mean | ± SD | |
| Hematocrit (%) | 38.60 | ± 3.10 | 37.50 | ± 10.20 |
| Erythrocytes (×1012/L) | 8.70 | ± 1.00 | 7.80 | ± 2.50 |
| Hemoglobin (g/dL) | 12.70 | ± 1.60 | 11.70 | ± 3.00 |
| MCV (fL) | 44.50 | ± 2.70 | 49.40 | ± 5.80 |
| MCHC (%) | 32.70 | ± 2.10 | 31.30 | ± 2.10 |
| WBC (×109/L) | 8.50 | ± 1.70 | 8.50 | ± 3.30 |
| PMN (×109/L) | 2.40 | ± 1.50 | 2.70 | ± 1.60 |
| Lymphocytes (×109/L) | 5.00 | ± 2.40 | 5.00 | ± 4.00 |
| Monocytes (×109/L) | 0.79 | ± 0.36 | 0.55 | ± 0.30 |
| Platelets (×109/L) | 939.00 | ± 65.00 | 1010.00 | ± 129.00 |
| Blood parameter | Donor PDGFRβ genotype | |||
|---|---|---|---|---|
| +/+ (n = 5) | −/− (n = 5) | |||
| Mean | ± SD | Mean | ± SD | |
| Hematocrit (%) | 38.60 | ± 3.10 | 37.50 | ± 10.20 |
| Erythrocytes (×1012/L) | 8.70 | ± 1.00 | 7.80 | ± 2.50 |
| Hemoglobin (g/dL) | 12.70 | ± 1.60 | 11.70 | ± 3.00 |
| MCV (fL) | 44.50 | ± 2.70 | 49.40 | ± 5.80 |
| MCHC (%) | 32.70 | ± 2.10 | 31.30 | ± 2.10 |
| WBC (×109/L) | 8.50 | ± 1.70 | 8.50 | ± 3.30 |
| PMN (×109/L) | 2.40 | ± 1.50 | 2.70 | ± 1.60 |
| Lymphocytes (×109/L) | 5.00 | ± 2.40 | 5.00 | ± 4.00 |
| Monocytes (×109/L) | 0.79 | ± 0.36 | 0.55 | ± 0.30 |
| Platelets (×109/L) | 939.00 | ± 65.00 | 1010.00 | ± 129.00 |
Peripheral blood parameters of hematopoietic chimeras were evaluated 14 to 18 weeks after transplantation. See Table 3 for abbreviations.
Identical reconstitution kinetics of wild-type and PDGF-B−/− hematopoietic cells after serial transplantation
We tested the possibility that stress induced by the transplantation of marrow from established chimeras into secondary recipients might reveal differences between PDGF-B+/+ and PDGF-B−/− hematopoietic stem cells. The second generation chimeras appeared healthy, and at 28 days after transplantation, the proportions of donor-derived B- and T-lymphoid and myeloid cells were comparable to those found after fetal liver transplantation (Figure4; compare with Figure 3A). No significant differences were observed in the reconstitution of wild-type and PDGF-B−/− second-generation chimeras (Figure 4).

Bone marrow from PDGF-B−/− chimeras can reconstitute the peripheral blood compartment of lethally irradiated recipients.
Marrow from first-generation PDGF-B+/+ and PDGF-B−/− chimeras was transplanted into lethally irradiated B6.Ly-5a recipients (PDGF-B+/+, n = 11; PDGF-B−/−, n = 12), and donor engraftment in second-generation chimeras was determined 28 days later.
Bone marrow from PDGF-B−/− chimeras can reconstitute the peripheral blood compartment of lethally irradiated recipients.
Marrow from first-generation PDGF-B+/+ and PDGF-B−/− chimeras was transplanted into lethally irradiated B6.Ly-5a recipients (PDGF-B+/+, n = 11; PDGF-B−/−, n = 12), and donor engraftment in second-generation chimeras was determined 28 days later.
Expression of PDGF-B and PDGFRβ in the fetal mouse liver
To address the possibility that the prominent reduction in size of PDGF-B−/− and PDGFRβ−/− fetal livers reflected a direct role of PDGF in the liver, we determined the expression patterns of PDGF-B and PDGFRβ in the fetal mouse liver by nonradioactive in situ hybridization. PDGF-B mRNA was strongly expressed by a subpopulation of cells in the developing liver (Figure 5A). These cells had the morphologic hallmarks of megakaryocytes—large and characterized by multilobulated nuclei (Figure 5B). In hematoxylin-eosin–stained sections, fetal livers contained comparable proportions of megakaryocytes in E16.5 to E18.5 PDGF-B+/+ and PDGF-B−/− livers (data not shown). We concluded that the only cell type with detectable PDGF-B mRNA expression in the fetal mouse liver was the megakaryocyte and that PDGF-B deficiency did not block megakaryocyte development in PDGF-B−/− embryos.

Limited expression of PDGF-B and PDGFRβ in E14.5 fetal mouse livers.
Nonradioactive in situ hybridization shows PDGF-B expression in a subset of liver cells (A, arrows), which have the morphologic hallmarks of megakaryocytes (B, arrow). PDGFRβ expression is low in the liver (C, Li), in comparison with the adjacent adrenal (C, Ad), in which abundant positive cells with perivascular location are seen (arrowheads). The developing hepatic connective tissue capsule expresses PDGFRβ, as shown in the homogeneous layer of stained cells between the liver tissue and the adrenal.
Limited expression of PDGF-B and PDGFRβ in E14.5 fetal mouse livers.
Nonradioactive in situ hybridization shows PDGF-B expression in a subset of liver cells (A, arrows), which have the morphologic hallmarks of megakaryocytes (B, arrow). PDGFRβ expression is low in the liver (C, Li), in comparison with the adjacent adrenal (C, Ad), in which abundant positive cells with perivascular location are seen (arrowheads). The developing hepatic connective tissue capsule expresses PDGFRβ, as shown in the homogeneous layer of stained cells between the liver tissue and the adrenal.
PDGFRβ expression in the fetal liver (Li) was restricted to the connective tissue capsule and to a low-abundance cell type of perivascular location in peripheral regions (eg, in the vicinity of the capsule) (Figure 5C, data not shown). This is in marked contrast to many other tissues, exemplified in Figure 5C by the adrenal (Ad), in which perivascular PDGFRβ-positive cells are abundant.10 11
We concluded that PDGF-B and PDGFRβ expression patterns were different in the liver than in many other embryonic tissues in that PDGF-B was not expressed by vascular endothelial cells and that PDGFRβ was not expressed by most developing pericytes. We previously reported that perisinusoidal pericytes (Ito cells) have normal abundances in PDGF-B and PDGFRβ−/− embryos, in contrast to many other types of pericytes.11 The reduced liver size in PDGF-B and PDGFRβ−/− embryos, therefore, was probably not a result of lack of PDGF-B/Rβ signaling within the liver.
Discussion
What role does PDGF play in hematopoiesis?
Previous in vitro and in vivo studies have suggested a role for PDGF in erythropoiesis17,18,20,21 and in stimulation of pluripotent stem cells.19,21,32-34 PDGF promotes in vitro proliferation of erythropoietic progenitors,20 but stimulation requires the presence of adherent stromal cells,21 presumably because of PDGF induction of colony-stimulating factor (CSF) release from the stromal cells. Mouse splenic erythroid cells express PDGF in vivo after stimulation with erythropoietin,17,18 demonstrating that precursors of the erythroid lineages are a source of PDGF. In mixed marrow cultures, PDGF stimulates the growth of primitive hematopoietic progenitor cells, such as colony-forming units containing granulocytes, erythroid cells, macrophages, megakaryocytes (CFU-mix, CFU-GEMM),19,32,33 and lineage-restricted hematopoietic progenitors.34 Adherent accessory cells are also required for the stimulation of primitive and lineage-restricted hematopoietic progenitors by PDGF, and growth enhancement appears to occur as an indirect effect through the stimulation of interleukin-1 (IL-1) production by PDGFR-positive stromal cells, including macrophages.19 All the above in vitro studies were performed with PDGF that contained PDGF-AA, -AB, and -BB. However, no specific effects of PDGF-AA or PDGF-CC have been reported. Overexpression of PDGF-B in murine hematopoietic cells induces a lethal myeloproliferative syndrome in vivo.35 Taken together, in vitro and in vivo studies suggest that PDGF enhances hematopoiesis by stimulating stromal cells to produce a variety of factors that act directly on hematopoietic progenitors.
Bone marrow stromal cells play a crucial role in sustaining proliferation, differentiation, and self-renewal of hematopoietic stem cells.36,37 It has also been shown that hematopoietic stem cells, not committed progenitor cells, mediate early hematopoietic reconstitution.38 Within the marrow, the principal sources of PDGF are megakaryocytes, platelets, erythroid precursors, and activated monocytes–macrophages.3 Populations known to express PDGF receptors are predominantly stromal cells, including fibroblasts, smooth muscle cells, osteoblasts, early macrophage precursors, and macrophages. Our finding that PDGF-B null mutant chimeras have a normal hematologic phenotype demonstrates that neither the release of PDGF from the hematopoietic cells nor the response of PDGFRβ-expressing hematopoietic cells to PDGF, such as the induction of IL-1 in receptor-expressing macrophages, is required for normal hematopoiesis in vivo. It is important to note that most stromal cells found in the marrow after lethal irradiation and marrow transplantation are derived from the recipient.37 39 Thus, in chimeras reconstituted with PDGFRβ−/− mutant stem cells, most stromal cells express PDGFRβ.
Our results with PDGF null hematopoietic chimeras contrast with the results of studies of mice with targeted deletion of growth factors known to stimulate hematopoiesis directly, such as erythropoietin,30,40 M-CSF,41 and thrombopoietin,42,43 which are required for the normal development of specific hematologic lineages. Some hematopoietic growth factors appear to serve redundant functions in hematopoiesis, as indicated by the observation that GM–CSF null mutant mice have normal hematopoietic parameters44,45 in spite of more subtle functional defects.46 Our results suggest that PDGF may also have a redundant function in hematopoiesis. Stromal production of hematopoietic cytokines can be stimulated not only by PDGF but also by other growth factors and cytokines, such as fibroblast growth factor, heparin-binding epidermal growth factor, epidermal growth factor, IL-1, and tumor necrosis factor-α.47-50 In keeping with our results, the targeted deletion of IL-1 and tumor necrosis factor-α did not produce any abnormalities in hematopoiesis.51-53 Whereas our experiments show that hematopoietic functions do not depend on expression of PDGF-B or PDGFRβ by fetal liver cells that completely repopulate the marrow, the data do not rule out the possibility that PDGF and other growth factors with overlapping activities contribute to normal hematopoiesis.54
Pathogenesis of disorders in PDGF-B– and PDGFRβ-deficient mice
Previous analysis of PDGF-B−/− embryos has demonstrated a severe pericyte deficiency in capillaries at many sites and smooth muscle hypoplasia in many larger vessels.10,11It is likely that the pericyte loss leads to the microaneurysms, tissue edema, and petechial hemorrhage characteristic of these mice. It is possible that the bleeding may contribute to the anemia seen in the mutants, but 2 observations suggest other principal causes of the anemia. First, anemia is present at E17.5, at which time most mutants have not yet developed significant hemorrhaging.7 Second, the anemia correlates with a consistent, approximately 50%, reduction in the size of the liver, the major hematopoietic organ at mid to late gestation. The size reduction is balanced (ie, normal ratios between the different cell types are preserved) and is apparent by E14.5, far earlier than the development of microaneurysms, tissue edema, and petechial hemorrhage. Vascular leakage leading to reduced fetal liver cellularity has also been observed in mice with null mutations in Tie1, a receptor tyrosine kinase essential for vascular cell integrity.55
The reduced liver size may be sufficient to explain the anemia and thrombocytopenia, but what is causing the reduction in liver size? Our in situ hybridization analysis demonstrates PDGF-B expression in megakaryocytes in E14.5 fetal livers, but PDGFRβ expression is not detected. In agreement with the lack of vascular expression of PDGF-B and PDGFRβ in the developing liver, PDGF-B and PDGFRβ have been shown to be dispensable for the development of liver pericytes (Ito cells).11 There also is no evidence for the positive selection of PDGFRβ-expressing cells in the livers of chimeric mice generated by fusing wild-type and PDGFRβ−/−embryos.56 Thus, the reduction in liver size is probably not coupled to deficient PDGF-B–PDGFRβ signaling within the livers but rather is secondary to defects elsewhere.
Two defects might be relevant to the reduction in liver size. First, PDGF-B and PDGFRβ−/− embryos show abnormal development of the placenta labyrinth that constitutes the fetal–maternal interface from E13.5 onward.12 As a result of dilated fetal vessels and reduced density of labyrinth trophoblasts in the mutants, the fetal–maternal interface is reduced in size.12 This likely produces metabolic stress, which could exert a particularly strong effect on rapidly growing embryonic tissues, such as the liver and the hematopoietic system. In p38α mitogen-activated protein (MAP) kinase knockouts that have a combined severe placental defect with trophoblast impairment and anemia and die at E13, there appears to be a placenta-to-liver axis that controls erythropoietin levels.57 Erythropoietin levels are decreased in the fetal livers of the p38α MAP kinase knockout embryos, and extra-embryonic tissue rescues the anemia. However, unlike the p38α MAP kinase knockout, we show that PDGF-B−/− embryos have increased erythropoietin levels compared with PDGF-B+/+ embryos. Second, heart dilation is observed from E13.5 in PDGF-B and PDGFRβ−/− embryos (M. Kalén, C. Betsholtz, unpublished observations, April 2000). This may reflect placental dysfunction, as proposed in PPARγ-deficient mice,58 but a dysfunctional heart may also lead to metabolic stress and hypoxia. As another sign of metabolic and hypoxic stress, vascular endothelial growth factor levels were increased in many PDGF-B and PDGFRβ−/− embryonic organs, including the liver (M. Hellström, C. Betsholtz, unpublished observations, January 2001). The increase in erythropoietin levels that we observed would be consistent with hypoxia-induced stress.
In summary, the hematopoietic chimera analysis, the normal embryonic PDGF-B and PDGFRβ expression patterns, and the combination of organ defects in mutants suggest that the hematologic disorders of PDGF-B– and PDGFRβ-deficient mice are linked to liver hypocellularity, which in turn results from systemic influences, possibly metabolic stress and hypoxia, caused by extrahepatic defects in these mutants.
PDGF-B−/− and PDGFRβ−/− hematopoietic chimeras: useful models to study inflammatory responses
The chimera model described in this study allows examination of marrow-derived cells in adult mice with targeted PDGF-B and PDGFRβ gene deletions that are otherwise lethal during gestation. After lethal total body irradiation, recipient granulocytes, monocytes, and B cells are replaced rapidly by donor-derived populations, whereas recipient T cells are replaced more slowly. We surmise that some T cells are resistant to irradiation-induced interphase death.59However, as judged by the absence of major histocompatibility complex–incompatible marrow graft rejection, it appears that most recipient T cells remaining after 14-Gy total body irradiation are functionally incompetent, presumably because they die whenever they are stimulated to replicate. Therefore, their persistence after transplantation probably reflects the long life span characteristic of certain T-cell populations.60 The slower repopulation of T cells in this model will have to be considered in experiments to test immune and inflammatory responses.
With the exception of the platelet, whose α granules contain PDGF, circulating blood cells normally express low or undetectable levels of PDGF.3 However, activation can induce PDGF synthesis in monocytes and erythroid cells, and activated platelets release PDGF from α granules. In several models of acute injury, use of blocking antibodies against PDGF significantly reduced subsequent connective tissue accumulation.3 However, the role of PDGF in chronic inflammatory responses cannot be easily studied with the use of neutralizing antibodies, unless administration of the antibody can be targeted to a narrow time window. The chimera model described in this report allows selective targeting of genes in circulating cells, a major source of PDGF in chronic inflammation, and can be applied to studies of other inflammatory genes. For PDGF biology, this model provides a system to investigate a variety of proposed functions inferred from in vitro studies or associative in vivo data. Issues that could be addressed with these chimeras include the role of PDGF-B and PDGFRβ in the immune response, specifically antigen-driven, T-cell proliferation, and natural killer cell cytotoxicity; the significance of PDGF in monocytes and platelets for the chronic inflammatory response in atherosclerosis and arthritis; and the function of PDGFRβ-dependent signaling in activation of monocyte-mediated inflammatory responses.
We thank Kelli McIntyre, Karen Engel, Roderick Browne, and Li-Chuan Huang for expert technical assistance, Philippe Soriano (Fred Hutchinson Cancer Research Center, Seattle, WA) for provision of PDGFRβ mutant mice, Thalia Papayannopoulou and Philippe Soriano for helpful discussions, and Barbara Droker for editorial assistance.
Supported by National Institutes of Health grants HL18645 (R.R., E.W.R.), HL55257 (P.J.M.), HL03174 (D.F.B.-P.), CA31615 (V.C.B.); Merck (R.R.); Swedish Medical Research Council, Cancer Foundation, Inga-Britt and Arne Lundberg Foundation, Novo Nordisk Foundation (C.B.); and Deutsche Forschungsgemeinschaft Ka 1078/1 (W.E.K.).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal