

Abstract
Most clinical trials with fibrinogen receptor antagonists (FRAs) have been associated with thrombocytopenia. This report describes the occurrence of thrombocytopenia in one chimpanzee and one rhesus monkey upon administration of potent FRAs. Chimpanzee A-264 experienced profound thrombocytopenia on two occasions immediately upon intravenous administration of two different potent FRAs, L-738,167 and L-739,758. However, an equally efficacious antiaggregatory dose of another potent antagonist, L-734,217, caused no change in platelet count. These compounds did not affect platelet count in five other chimpanzees or numerous other nonhuman primates. Flow cytometric analysis showed drug-dependent antibodies (DDAbs) in the plasma of chimpanzee A-264 that bound to platelets of chimpanzees, humans, and all other primates tested only in the presence of the compounds that induced thrombocytopenia. Rhesus monkey 94-R021 experienced thrombocytopenia upon administration of a different antagonist, L-767,679, and several prodrugs that are converted into the active form, L-767,679, in the blood. More than 20 other FRAs, including those that induced thrombocytopenia in chimpanzee A-264, had no effect on platelet count in this monkey. Flow cytometric measurements again identified DDAbs that reacted with platelets of all primates tested and required the presence of L-767,679. Screening for DDAbs in the plasma of 1,032 human subjects with L-738,167 and L-739,758 demonstrated that the incidence of these preexisting antibodies in this population was 0.8% ± 0.6% and 1.1% ± 0.6%, respectively.
RECENT CLINICAL TRIALS with platelet fibrinogen receptor antagonists (FRAs) demonstrated that glycoprotein (GP) IIb/IIIa blockade represents a major new advancement in the management of patients with coronary disease.1-3 However, all clinical trials, with one exception,4 have consistently been associated with an incidence of thrombocytopenia (typically 1% to 2% of patients).3-10 The observed thrombocytopenias have been of two types: immediate, within hours, or with a delayed onset, several days after drug administration. Although clinical thrombocytopenia is defined as a platelet count of less than 100,000 platelets/μL and severe thrombocytopenia is defined as a platelet count of less than 50,000 platelets/μL, some cases associated with GP IIb/IIIa antagonist administration have been profound (<20,000 platelets/μL).5-7 The mechanism underlying profound thrombocytopenia after the initial exposure to FRAs, occurring in some cases within several hours after administration of a GP IIb/IIIa blocker,6,7 is poorly understood.3 5
Drug-induced thrombocytopenia was described as early as 1928 during quinine therapy.11 Since that time, immune-mediated thrombocytopenia associated with drug-dependent antibodies (DDAbs) has been well documented for quinine, quinidine, and more than 100 other low molecular weight compounds as well as currently used therapeutics.5,11-15 The mechanism of immune-mediated thrombocytopenia remains unknown5; however, it has been frequently demonstrated that DDAbs recognize epitopes exposed on the drug-receptor complex upon binding of the drug.5Interestingly, epitopes for quinine/quinidine-dependent antibodies described first on the platelet GP Ib/IX complex16,17 were later also found on the GP IIb/IIIa complex.18-20 Recently, antigens on GP IIb/IIIa have been identified as targets of DDAbs in patients who experienced pentamidine- and sulfonamide-induced thrombocytopenia.14 15
Numerous methods have been developed for the detection of platelet-associated DDAbs in patients with drug-induced thrombocytopenia.5,15,21-23 The recently described flow cytometric method15 provides a very sensitive technique for detection of antiplatelet antibodies using whole platelets. This assay closely mimics the conditions in whole blood, without significantly disturbing neoepitopes exposed on the platelet surface upon drug administration.15
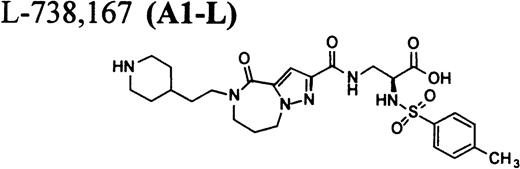
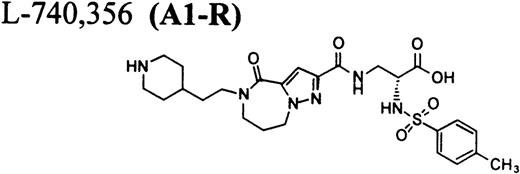
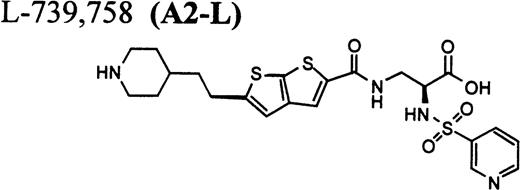
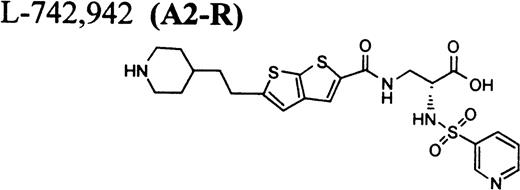
In this report, we describe thrombocytopenia in two nonhuman primates induced by the administration of structurally and functionally diverse RGD-mimetic, FRAs: L-738,167-(A1-L),24,25L-739,758-(A2-L),26 and L-767,679-(B).27 We also provide direct flow cytometric evidence that the plasma of both primates contains preexisting DDAbs that recognize epitopes on GP IIb/IIIa of all human and nonhuman primates evaluated. Screening for DDAbs in the plasma of 1,032 human subjects with L-738,167 and L-739,758 demonstrated that the incidence of these preexisting antibodies in this population was 0.8% ± 0.6% and 1.1% ± 0.6%, respectively.
MATERIALS AND METHODS
Experimental protocol for in vivo studies in chimpanzees, baboons, rhesus monkeys, cynomolgus monkeys, African green monkeys, squirrel monkeys, and dogs.
Chimpanzees were sedated with ketamine (10 mg/kg intramuscularly [IM]) for compound administration and blood withdrawal for the determination of platelet count and evaluation of platelet aggregation responses. Before intravenous (IV) bolus (10 μg/kg) administration of A1-L (n = 6) and A2-L (n = 1) or IV infusion (0.4 μg/kg/min for 60 minutes) of A3 (n = 4), a blood sample (sodium citrate; final concentration, 0.38%) was taken by venipuncture from a saphenous vein. This was used for determination of baseline whole blood platelet count and ex vivo platelet aggregation response to adenosine diphosphate (ADP). Additional blood samples were taken at selected time points after the initiation of treatment. Other nonhuman primates (squirrel monkey [n = 12], African green monkey [n = 4], rhesus monkey [n = 88], cynomolgus monkey [n = 79], and baboon [n = 12]) were also evaluated for FRA-induced thrombocytopenia. The studies with these primates were conducted at several different facilities and were, therefore, studied in either the conscious or sedated state depending on the normal procedures at each site. Conscious animals were placed in primate restraint chairs and transported to the laboratory on the day of the experiment; sedated animals were chemically restrained with ketamine for blood withdrawal and compound administration. Blood samples were taken before compound administration and at selected time points after initiation of treatment and used for platelet count and platelet aggregation measurements. A subpopulation of rhesus monkeys (n = 5) received A1-L after prior treatment with A3 (0.4 μg/kg/min for 60 minutes). A1-L was administered to conscious (n = 157) and anesthetized (n = 128, 35 mg/kg IV sodium pentobarbital) purpose-bred mongrel dogs of either sex. Conscious dogs rested comfortably in nylon slings for administration of A1-L orally (by gastric lavage in aqueous solution or as solid, crystalline material in capsule) or as an IV bolus and/or continuous infusion.
Measurement of ex vivo platelet aggregation responses and whole blood platelet count.
Platelet-rich plasma (PRP) was prepared by centrifugation of whole blood at 150g for 5 minutes and the platelet concentration was adjusted to 2 × 108 platelets/mL with time-matched platelet-poor plasma (PPP). PRP (300 μL, 2 × 108platelets/mL) was incubated at 37°C for 3 minutes before the addition of agonist. Platelet aggregation was measured by the change in light transmittance (PPP represents 100%) under stirring conditions (1,100 rpm) at 37°C in a Biodata Platelet Aggregation Profiler (Model PAP-4; Biodata Corp, Horsham, PA) and was initiated by the addition of 20 μmol/L ADP for primates and 10 μmol/L ADP + 1 μmol/L epinephrine for dogs. The extent of aggregation was taken as the peak percentage of aggregation achieved based on a maximum of 100%, and the effect of treatment on platelet aggregation was expressed as the percentage of inhibition using the baseline, pretreatment aggregation response as 100%. Whole blood platelet counts were determined using an automated hematology analyzer (Biochem Immunosystems, Allentown, PA) for all experiments except chimpanzees (Coulter Cell Counter; Coulter, Hialeah, FL).
Preparation of gel-filtered platelets (GFP).
GFP were prepared from blood anticoagulated with 13 mmol/L trisodium citrate. PRP was generated by centrifugation at 180g for 20 minutes in the presence of 1 μmol/L prostaglandin E1 (PGE1; Sigma Chemical, St Louis, MO). Platelets were separated from plasma by gel filtration through a Sepharose 2B column (Pharmacia Biotech, Uppsala, Sweden) and equilibrated in platelet buffer (138 mmol/L NaCl, 6 mmol/L KCl, 1.7 mmol/L Na2HPO4, 6.3 mmol/L HEPES, 1 mg/mL dextrose, and 2% bovine serum albumin [BSA], pH 7.4). The platelets were diluted to 2 × 108 cells/mL in platelet buffer before use.
RGD-mimetics.
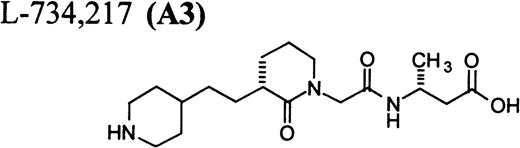
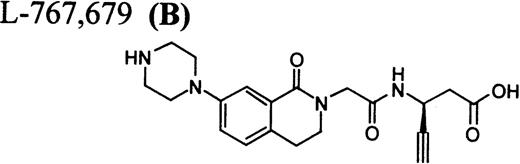
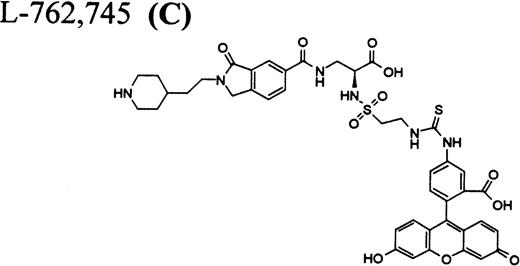
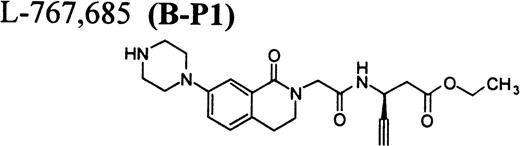
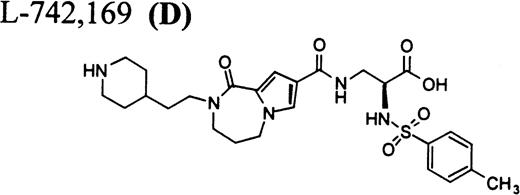
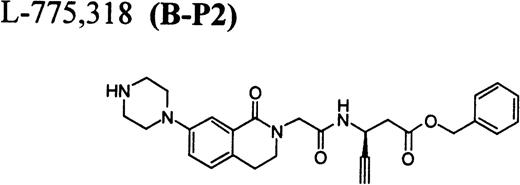
Flow cytometric assay of DDAbs.
The flow cytometric assay for detection of DDAbs is based on the method of Curtis et al.15 Platelets at a concentration of 1 × 108 platelets/mL in flow cytometric buffer (10 mmol/L HEPES, 5 mmol/L KCl, 145 mmol/L NaCl, and 1 mmol/L MgCl2) were incubated with platelet inhibition cocktail that contained PGI2 (10 μmol/L), apyrase (2 U/mL), leupeptin (0.5 mmol/L), and hirudin (10 nmol/L) and either FRAs (10 μmol/L) or buffer (control) for 15 minutes at room temperature. Platelet suspension (50 μL) was spun at 100g for 4 minutes and platelets resuspended in 100 μL of plasma containing platelet inhibition cocktail with or without FRA. The mixture was incubated for 1 hour at room temperature. The platelets were then pelleted (PGE1 added) as described previously and washed with 500 μL of flow cytometric buffer in the presence or absence of the appropriate FRA (10 μmol/L). The platelets were resuspended in 100 μL of flow cytometric buffer in the presence or absence (control) of FRA and 20 μL of platelet suspension was added to tubes containing 200 μL of goat fluorescein isothiocyanate (FITC)-labeled antihuman or antimonkey IgG (experiments with rhesus monkey plasma; Accurate Chemical & Scientific Co, Westbury, NY) and incubated for 1 hour at room temperature. The amount of bound FITC-antihuman or antimonkey IgG was determined by flow cytometry15 (FACSCalibur; Becton Dickinson, San Jose, CA).
Saturation of platelet receptors with DDAbs by repeat incubation with chimpanzee A-264 plasma.
Gel-filtered platelets (200 μL at 2 × 108platelets/mL) were incubated with FRA for 15 minutes as described previously. Platelets were pelleted and then resuspended in 200 μL of chimpanzee plasma containing platelet inhibition cocktail and incubated for 30 minutes at room temperature. The platelets were then either pelleted and resuspended with 200 μL of chimpanzee plasma again or washed with 500 μL of flow cytometric buffer and analyzed in the flow cytometer for DDAbs as previously described.
Enzyme-linked immunosorbent assay (ELISA) measurements.
Microtiter plate wells were coated overnight at 4°C with 5 μg/mL of purified GP IIb/IIIa29 diluted in phosphate-buffered saline (PBS) either in the presence or absence of 10 μmol/L A2-L. The wells were washed four times with PBS containing 0.05% Tween 20 (PBS/Tween 20) and incubated for 1 hour with either 100 μL of chimpanzee A-264 or control chimpanzee plasma diluted 1:20 in PBS/Tween 20 in the presence or absence of 10 μmol/L A2-L. The wells were then washed four times with PBS/Tween 20 and incubated with sheep antihuman IgG-peroxidase conjugate (Amersham, Cleveland, OH) diluted 1:1,000 in PBS/Tween 20 for 1 hour at room temperature. After washing the wells four times with PBS/Tween 20, the ABST peroxidase substrate (Kirkeqaard & Perry, Gaithersburg, MD) was added and the change in absorbance at 405 nm was measured after 15 minutes.
Screening for DDAbs in human plasma samples.
Human plasma samples (1,032 different anonymous blood donor subjects) were screened for A1-L– and A2-L–dependent antibodies using the previously described flow cytometric assay and human gel-filtered platelets from healthy donors. A plasma sample from chimpanzee A-264 was used as a positive control, and plasma samples from a control chimpanzee and a human subject (negative for DDAbs) were used as negative controls. The values of relative fluorescence (fluorescence of plasma in the presence of drug/fluorescence in the absence of drug) were 1.02 ± 0.17 and 1.05 ± 0.15 (mean ± SD, n = 60) for control chimpanzee plasma and control human plasma, respectively. Samples were considered positive when the value of relative fluorescence was ≥2, similar to Curtis et al.15 Whenever the plasma sample was found positive in the presence of either A1-L or A2-L, it was tested again using these two compounds and two other FRAs, A3 and B, on platelets from three different human donors. A plasma sample was reported positive if it was found positive using at least two different platelet donors. Positive samples tested positive on platelets of all tested platelet donors; however, the amount of detected bound DDAbs, as measured by the fluorescence ratio, varied for different platelet donors. We repeated testing of 200 samples that were found to be negative in the first evaluation. All of these samples remained negative with repeat testing.
RESULTS
Effect of administration of FRAs on platelet count in nonhuman primates.
One of six chimpanzees (A-264) experienced immediate profound thrombocytopenia upon IV bolus infusion of 10 μg/kg of a potent FRA, A1-L.24,25 As shown in Fig 1A, platelet aggregation was completely inhibited in all six chimpanzees (including A-264) for approximately 12 hours after IV administration of 10 μg/kg of A1-L. The antiaggregatory effect gradually decreased from 12 to 56 hours after A1-L treatment. As shown in Fig 1B, the platelet count for chimpanzee A-264 began to decrease (153,000, 132,000, 119,000, and 84,000 platelets/μL at baseline, 5, 15, and 30 minutes after treatment, respectively) immediately upon drug administration and reached a minimum (<40,000 platelets/μL) at 2 hours after dosing. The platelet count remained at that level for the next 6 hours and then gradually increased from 10 to 96 hours (108,000 platelets/μL). The increase of platelet count was practically linear, with a rate consistent with the life span of platelets of about 6 to 7 days. The apparently small reduction in the initial platelet count in the five control chimpanzees is within experimental error of the measurement, although the trend seems to be similar for all animals (Fig 1B). This observation could be procedural or physiologically based; however, we have no direct evidence to support either. After full recovery, and more than 30 days after administration of A1-L, chimpanzee A-264 received an infusion of 0.4 μg/kg/min for 60 minutes of another FRA, A3.28,30 As shown in the Fig 2, A3 inhibited ex vivo platelet aggregation during 1 hour of infusion and for 2 hours after the end of infusion; however, there was no effect on platelet count. By 24 hours after treatment with A3, ex vivo platelet aggregation had returned to normal (Fig 2A). Chimpanzee A-264 was then challenged with an IV bolus of 10 μg/kg of A2-L, an FRA26functionally and structurally similar to A1-L. As shown in Fig 2B, the platelet count of chimpanzee A-264 decreased precipitously within 1 hour to less than 20,000 platelets/μL and then recovered with a similar time course to the recovery after treatment with A1-L. Chimpanzee A-264 has not been challenged with any other FRA since these thrombocytopenic episodes.

The effect of 10 μg/kg IV bolus of A1-L on ex vivo platelet aggregation in PRP stimulated with 20 μmol/L ADP (A) and platelet count in whole blood (B) in ketamine-sedated chimpanzees. (▴) Inhibition of platelet aggregation as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler for chimpanzee A-264 and five control chimpanzees (mean ± SEM, n = 6). Platelet count measured in whole blood using a Coulter Cell Counter for (•) control chimpanzees (mean ± SEM, n = 5) and (▪) chimpanzee A-264.
The effect of 10 μg/kg IV bolus of A1-L on ex vivo platelet aggregation in PRP stimulated with 20 μmol/L ADP (A) and platelet count in whole blood (B) in ketamine-sedated chimpanzees. (▴) Inhibition of platelet aggregation as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler for chimpanzee A-264 and five control chimpanzees (mean ± SEM, n = 6). Platelet count measured in whole blood using a Coulter Cell Counter for (•) control chimpanzees (mean ± SEM, n = 5) and (▪) chimpanzee A-264.

The effect of the infusion of A3 (0.4 μg/kg/min for 1 hour) followed 24 hours later by 10 μg/kg IV bolus of A2-L on ex vivo platelet aggregation in PRP stimulated with 20 μmol/L ADP (A) and platelet count in whole blood (B) in ketamine-sedated chimpanzee A-264. (•) Inhibition of platelet aggregation as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. (▪) Platelet count measured in whole blood using a Coulter Cell Counter.
The effect of the infusion of A3 (0.4 μg/kg/min for 1 hour) followed 24 hours later by 10 μg/kg IV bolus of A2-L on ex vivo platelet aggregation in PRP stimulated with 20 μmol/L ADP (A) and platelet count in whole blood (B) in ketamine-sedated chimpanzee A-264. (•) Inhibition of platelet aggregation as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. (▪) Platelet count measured in whole blood using a Coulter Cell Counter.
Part of our overall strategy for the investigation of the mechanism of thrombocytopenia induced by GP IIb/IIIa inhibitors as well as for assessing the risk for thrombocytopenia with other compounds was the identification of other primates susceptible to thrombocytopenia upon exposure to a potent FRA (A1-L). As described in the methods, A1-L was administered as an IV bolus of 10 μg/kg to numerous primate species, including chimpanzee (n = 6), rhesus monkey (n = 88), cynomolgus monkey (n = 79), African green monkey (n = 4), squirrel monkey (n = 12), and baboon (n = 12). The whole blood platelet count at 2 and 4 hours after dosing did not decrease to the level of thrombocytopenia in any animal (with or without pretreatment with A3) except chimpanzee A-264. Therefore, thrombocytopenia occurred in 0.5% ± 0.5% of the primates in this population (1/201).
Binding characteristics of selected FRAs in binding to chimpanzee and human platelets.
Because all described FRAs are highly selective24-28ligands for GP IIb/IIIa versus other integrins, it was of interest to compare their binding affinities to GP IIb/IIIa on platelets from chimpanzee A-264, a control chimpanzee, and human platelets. As shown in Table 2, dissociation constants of the fluorescein-labeled FRA31 32 C as well as A1-L and A2-L in binding to chimpanzee A-264, control chimpanzee, and human platelets are identical within experimental error. We also considered the possibility that the unique response in chimpanzee A-264 versus other primates could be due to a dissimilar number of receptors per platelet. However, the number of GP IIb/IIIa receptors per platelet was found identical for both chimpanzees and within the range found for 100 human subjects (Table 2). We further found, using flow cytometry, that the size as well as the distribution in the size of platelets for chimpanzee A-264 and control chimpanzee was identical and comparable with data for human platelets (data not shown). The binding of FRA (used in our report) to the receptors on resting platelets does not result in the activation of platelets as measured by standard methods (calcium flux, serotonin, and P-selectin secretion), although all compounds induce more ligand-induced binding sites (LIBS) and expansion upon binding to GP IIb/IIIa (data not shown).
Binding Characteristics of FRAs and Number of GP IIb/IIIa Receptors Per Platelet on Chimpanzee A-264, Control Chimpanzee, and Human Platelets
| Species | Kd (nmol/L) | koff (×104/s−1) (A1-L) | No. of Receptors Per Platelet (×10−3) | ||
|---|---|---|---|---|---|
| C | A1-L | A2-L | |||
| Chimpanzee A-264 | 16 ± 3 | 0.30 ± 0.11 | 0.08 ± 0.03 | 5.2 ± 0.4 | 78 ± 14 |
| Control chimpanzee | 15 ± 3 | 0.28 ± 0.12 | 0.09 ± 0.04 | 5.1 ± 0.3 | 72 ± 12 |
| Human platelets | 15 ± 6 | 0.33 ± 14 | 0.10 ± 0.05 | 4.6 ± 0.4 | 57 ± 12* |
| Species | Kd (nmol/L) | koff (×104/s−1) (A1-L) | No. of Receptors Per Platelet (×10−3) | ||
|---|---|---|---|---|---|
| C | A1-L | A2-L | |||
| Chimpanzee A-264 | 16 ± 3 | 0.30 ± 0.11 | 0.08 ± 0.03 | 5.2 ± 0.4 | 78 ± 14 |
| Control chimpanzee | 15 ± 3 | 0.28 ± 0.12 | 0.09 ± 0.04 | 5.1 ± 0.3 | 72 ± 12 |
| Human platelets | 15 ± 6 | 0.33 ± 14 | 0.10 ± 0.05 | 4.6 ± 0.4 | 57 ± 12* |
The values of kd and koff were determined as described in Bednar et al.32 Mean ± SD of at least three measurements.
Average of measurements in 100 human subjects.
Flow cytometric measurement of DDAbs in plasma from chimpanzee A-264.
It is also possible that the observed thrombocytopenia in chimpanzee A-264 could be mediated by an immune response. To determine whether plasma from chimpanzee A-264 contained drug-dependent antiplatelet antibodies, we used a slightly modified flow cytometric method described by Curtis et al15 (for details, see Materials and Methods). Figure 3A shows histograms from the flow cytometric measurements of DDAbs using plasma and platelets from chimpanzee A-264 and A2-L. The shape of the flow cytometric histogram in Fig 3 (+ A2-L) is indistinguishable from the histogram obtained by size measuring light scattering detector (not shown), indicating that DDAbs bind equally well to the receptors on all platelets present. Figure 3B depicts results of flow cytometric measurements on chimpanzee A-264 and control chimpanzee plasma using platelets from both animals. As shown in Fig 3B, DDAbs from chimpanzee A-264 plasma selectively reacted with platelets from both chimpanzees (control or A-264) in the presence of A1-L and A2-L. However, the amount of antibody bound to platelets as indicated by the relative fluorescence was significantly lower for A1-L in all measurements. No DDAbs were detected in the presence of A3, an FRA that did not have an effect on platelet count in chimpanzee A-264. Similar results were obtained when, instead of chimpanzee platelets, platelets from 45 human subjects (see Fig 3C) and several other nonhuman primates (data not shown) were used in incubation with chimpanzee plasma. Thus, chimpanzee A-264 plasma appears to contain DDAbs that recognize an epitope on chimpanzee, human, and several other nonhuman primate platelets.



Flow cytometric measurement of antibody binding from chimpanzee A-264 and control chimpanzee plasma alternatively using chimpanzee A-264 and control chimpanzee platelets (A and B) and human platelets (C) in the presence of 10 μmol/L drug. (A) depicts the histograms from flow cytometric measurements of DDAb binding in the presence (+) and in the absence (−) of A2-L. Data in (B) represent the mean ± SD from three measurements. Data in (C) using A2-L and A1-L represent the mean ± SD from measurements using platelets of 45 human subjects; for other compounds, data represent the mean ± SD from measurements using platelets of three human subjects. Binding of DDAbs is plotted as relative fluorescence of an FITC-labeled secondary antibody. The right two sets of bars in (C) represent results with human platelets pretreated at 37°C for 1 hour with 10 mmol/L EDTA. A2-R (Kd, 0.45 nmol/L) and A1-R (Kd, 0.28 nmol/L) are R-isomers of A2-L and A1-L, respectively.
Flow cytometric measurement of antibody binding from chimpanzee A-264 and control chimpanzee plasma alternatively using chimpanzee A-264 and control chimpanzee platelets (A and B) and human platelets (C) in the presence of 10 μmol/L drug. (A) depicts the histograms from flow cytometric measurements of DDAb binding in the presence (+) and in the absence (−) of A2-L. Data in (B) represent the mean ± SD from three measurements. Data in (C) using A2-L and A1-L represent the mean ± SD from measurements using platelets of 45 human subjects; for other compounds, data represent the mean ± SD from measurements using platelets of three human subjects. Binding of DDAbs is plotted as relative fluorescence of an FITC-labeled secondary antibody. The right two sets of bars in (C) represent results with human platelets pretreated at 37°C for 1 hour with 10 mmol/L EDTA. A2-R (Kd, 0.45 nmol/L) and A1-R (Kd, 0.28 nmol/L) are R-isomers of A2-L and A1-L, respectively.
It is interesting that the R-isomer of A2-L (compound A2-R), which is also a potent FRA, induced binding of DDAbs to human platelets similar to A2-L, whereas the R-isomer of A1-L (A1-R) had no effect (Fig 3C). Because the concentration of FRAs used in the flow cytometric assay (10 μmol/L; ∼10-fold higher than the maximum in vivo concentration) is much higher than the possible maximal concentration of receptors (∼10 nmol/L) and higher than the dissociation constants (see Table 2), all GP IIb/IIIa receptors on the surface of platelets can be assumed to be occupied by FRAs. Results of flow cytometric measurements of DDAbs presented in Fig 3 were obtained with FITC-labeled goat antihuman IgG secondary antibody. When goat antihuman IgM or IgA secondary antibodies were used, no DDAbs in chimpanzee A-264 were detected (data not shown). Treatment of platelets with 10 mmol/L EDTA at 37°C causes dissociation of the GP IIb/IIIa complex on platelets, resulting in a complete loss33 34 of RGD-ligand binding as well as the dissociation of complex or conformational dependent antibodies. Flow cytometric measurements on human platelets (Fig 3C) indicated that the reaction of DDAbs from chimpanzee A-264 plasma with human platelets was abolished when the platelets were treated for 1 hour with 10 mmol/L EDTA at 37°C. An activation of platelets by either ADP or TRAP before incubation with plasma from chimpanzee A-264 did not induce binding of DDAbs (data not shown).
Both the plasma concentration of DDAbs as well as the affinity of the antibodies for platelets may play critical roles in the induction of thrombocytopenia. Flow cytometric measurements with different dilutions of chimpanzee A-264 plasma indicated that, upon dilution of plasma to one fifth the fluorescence as a measure of platelet-bound antibodies decreased approximately to the level found with control chimpanzee plasma. Flow cytometric measurement of DDAbs in chimpanzee A-264 plasma collected over 30 months showed that the titer of DDAbs remained constant. Figure 4 shows that the fluorescence, as a measure of bound DDAbs in flow cytometric measurements, increased with increasing concentration of A2-L from 1 to 100 nmol/L and then remained constant. Results presented in Fig4 were obtained with GFP from a single human donor with 2 × 108 cells/mL and an average of 105,000 GP IIb/IIIa receptors per platelet, suggesting a concentration of receptors of about 35 nmol/L. The Kd value for the A2-L in binding to GP IIb/IIIa on platelets is approximately 0.1 nmol/L (see Table 2); therefore, most of the receptors should be occupied at equilibrium when the concentration of the drug reaches the concentration of receptors (∼35 nmol/L). Thus, the maximum binding of DDAbs should be reached at an A2-L concentration of 35 nmol/L. However, during the wash step of the experimental procedure, equilibrium is not maintained. Thus, the receptor occupancy is always lower than it would be at equilibrium conditions. This results in a shift of the concentration necessary for full receptor occupancy to higher concentrations. As a result, the maximum fluorescence in Fig 4 is shifted also to higher concentrations of A2-L, whereas at the concentration 35 nmol/L, approximately 90% of DDAb is bound. Data in Fig 4 further indicate that the drug structure is not likely a major part of the epitope, because more than 100-fold excess of the free drug over the receptors does not lower DDAb binding.

The effect of the concentration of A2-L on the binding of DDAbs from chimpanzee A-264 plasma to human GFP (100 μL, 2 × 108 platelets/mL) as measured by the fluorescence of the FITC-labeled secondary antibody using flow cytometry. The concentration of GP IIb/IIIa receptors was 35 nmol/L.
The effect of the concentration of A2-L on the binding of DDAbs from chimpanzee A-264 plasma to human GFP (100 μL, 2 × 108 platelets/mL) as measured by the fluorescence of the FITC-labeled secondary antibody using flow cytometry. The concentration of GP IIb/IIIa receptors was 35 nmol/L.

The effect of repeated incubations of chimpanzee A-264 (A) and human platelets (B) with chimpanzee A-264 plasma on binding of DDAbs in the presence of 10 μmol/L drug as measured by relative fluorescence of FITC-labeled secondary antibody using flow cytometry. (A) One hundred microliters of 2 × 108 platelets/mL was repeatedly incubated with 60 μL of chimpanzee A-264 plasma. (▪) A2-L, (•) A1-L using chimpanzee A-264 plasma; (▴) A2-L, (▿) A1-L using control chimpanzee plasma. (B) Fifty microliters of 1 × 108 platelets/mL was repeatedly incubated with 100 μL of chimpanzee A-264 plasma. (▪) A2-L, (•) A1-L using chimpanzee A-264 plasma; (▴) A2-L using control chimpanzee plasma.
The effect of repeated incubations of chimpanzee A-264 (A) and human platelets (B) with chimpanzee A-264 plasma on binding of DDAbs in the presence of 10 μmol/L drug as measured by relative fluorescence of FITC-labeled secondary antibody using flow cytometry. (A) One hundred microliters of 2 × 108 platelets/mL was repeatedly incubated with 60 μL of chimpanzee A-264 plasma. (▪) A2-L, (•) A1-L using chimpanzee A-264 plasma; (▴) A2-L, (▿) A1-L using control chimpanzee plasma. (B) Fifty microliters of 1 × 108 platelets/mL was repeatedly incubated with 100 μL of chimpanzee A-264 plasma. (▪) A2-L, (•) A1-L using chimpanzee A-264 plasma; (▴) A2-L using control chimpanzee plasma.
The overall fluorescence intensity of the secondary FITC-labeled antihuman antibody in the flow cytometric assay was threefold to fivefold lower than expected if all GP IIb/IIIa receptors were occupied with DDAbs. Therefore, it was of interest to determine if either repeated incubation of the same platelets with chimpanzee A-264 plasma or incubation of the same number of platelets with a higher volume of plasma would result in an increase of the DDAb signal. As shown in Fig5A, when the original 100 μL of chimpanzee A-264 platelets was repeatedly incubated with 60 μL of chimpanzee A-264 plasma, the relative fluorescence of the secondary antibody increased and leveled off after eight incubations. This indicates a roughly fourfold increase in the amount of bound A2-L–dependent antibody when compared with a single incubation. Interestingly, in a parallel experiment with A1-L, relative fluorescence was unaffected after repeated incubations with fresh chimpanzee A-264 plasma. A similar experiment using 50 μL of human platelets in repetitive incubations with 100 μL of chimpanzee A-264 plasma (Fig 5B) showed that the maximum bound A2-L–dependent antibody was reached after four incubations. This finding agrees well with the previous experiment with chimpanzee A-264 platelets, because the amount of platelets in the experiment with human platelets was one half, and it was incubated with 100 μL of plasma instead of 60 μL used in the experiment with chimpanzee A-264 platelets. Thus, saturation of receptors by DDAbs was achieved after four incubations instead of eight with chimpanzee A-264 platelets. When the plasma of chimpanzee A-264 that was used in the experiment was used for a second time on fresh platelets, no DDAbs were found. Similar to the experiments with chimpanzee A-264 platelets, repetitive incubation of human platelets with chimpanzee A-264 plasma had little effect on the amount of bound A1-L–dependent antibodies. Repeated incubations of platelets with control chimp plasma had no effect on relative fluorescence, indicating that there were no DDAbs present.
FRA-induced thrombocytopenia in rhesus monkey 94-R021.
After testing numerous FRAs in various animal species (including 285 dogs dosed with A1-L alone), we identified only one primate besides chimpanzee A-264 that experienced FRA-induced thrombocytopenia. After oral administration of 0.3 mg/kg of B-P1 and 0.5 mg/kg of B-P2, rhesus monkey 94-R021 showed complete inhibition of platelet aggregation (Fig6A) and an approximately 40% to 80% decrease in platelet count, respectively (Fig 6B). These compounds are both ester prodrugs that convert to the active metabolite B27 in the blood. The time course of the onset of thrombocytopenia in the rhesus monkey correlates with the conversion of prodrugs to the active acid B. To further characterize the thrombocytopenic response, this monkey received B as an IV bolus of 20 μg/kg. Upon administration of B, this rhesus monkey demonstrated a precipitous decrease in platelet count (517,000 platelets/μL baseline to 144,000 platelets/μL at 30 minutes postdose; Fig 6C). However, in contrast to chimpanzee A-264, the platelet count in rhesus monkey 94-R021 started to increase at 60 minutes (187,000 platelets/μL) after dosing B and nearly recovered (441,000 platelets/μL) within 6 hours (Fig 6C). The recovery of the platelet count started when the receptor occupancy by B (see Fig 6D) decreased to less than approximately 40% (40,000) of the total GP IIb/IIIa receptors. When rhesus monkey 94-R021 was challenged with 5 μg/kg IV bolus of A2-L, which completely inhibited platelet aggregation for several hours and induced thrombocytopenia in chimpanzee A-264, no change in platelet count was observed (Fig 6C). Similarly, 20 other FRAs (including A1-L and A3) that completely inhibited platelet aggregation had no effect on platelet count in 94-R021 (data not shown). When rhesus monkey 94-R021 was dosed with 10 μg/kg IV bolus of the FRA D, which is structurally and functionally similar to A1-L, platelet aggregation was completely inhibited with no effect on platelet count (Fig7). Interestingly, the administration of B after the administration of D had no effect on platelet count. Thus, the binding of D to GP IIb/IIIa on rhesus monkey 94-R021 platelets prevented thrombocytopenia otherwise induced by the administration of B.
![Fig. 6. The effect of 0.5 mg/kg (administered orally) of B-P2 (phenyl ester of B) and 0.3 mg/kg (administered orally) of B-P1 (ethyl ester of B) on ex vivo platelet aggregation in PRP upon stimulation with 20 μmol/L ADP (A) and platelet count in whole blood (B) in rhesus monkey 94-R021. The effect of 5 μg/kg IV bolus of A2-L and 20 μg/kg IV bolus of B on ex vivo platelet aggregation in PRP upon stimulation with 20 μmol/L ADP and platelet count in whole blood (C) and number of receptors occupied by B (D) in rhesus monkey 94-R021. (A and B) Inhibition of platelet aggregation by B-P1 (▾) or B-P2 (▴) as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. Platelet count measured in whole blood of rhesus monkey 94-R021 using an automated hematology analyzer (Biochem Immunosystems) upon dosing B-P1 (•) and B-P2 (▪). (C and D) Platelet count measured in whole blood of rhesus monkey 94-R021 using an automated hematology analyzer (Biochem Immunosystems) upon dosing A2-L (•) and B (▪). Inhibition of platelet aggregation by B (▾) or A2-L (⧫) as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. The number of GP IIb/IIIa receptors occupied by B as measured by competition displacement measurements with [125I]-L-692,884 (▴).](/view-large/figure/9115363/blod41428006x.jpeg)
The effect of 0.5 mg/kg (administered orally) of B-P2 (phenyl ester of B) and 0.3 mg/kg (administered orally) of B-P1 (ethyl ester of B) on ex vivo platelet aggregation in PRP upon stimulation with 20 μmol/L ADP (A) and platelet count in whole blood (B) in rhesus monkey 94-R021. The effect of 5 μg/kg IV bolus of A2-L and 20 μg/kg IV bolus of B on ex vivo platelet aggregation in PRP upon stimulation with 20 μmol/L ADP and platelet count in whole blood (C) and number of receptors occupied by B (D) in rhesus monkey 94-R021. (A and B) Inhibition of platelet aggregation by B-P1 (▾) or B-P2 (▴) as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. Platelet count measured in whole blood of rhesus monkey 94-R021 using an automated hematology analyzer (Biochem Immunosystems) upon dosing B-P1 (•) and B-P2 (▪). (C and D) Platelet count measured in whole blood of rhesus monkey 94-R021 using an automated hematology analyzer (Biochem Immunosystems) upon dosing A2-L (•) and B (▪). Inhibition of platelet aggregation by B (▾) or A2-L (⧫) as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. The number of GP IIb/IIIa receptors occupied by B as measured by competition displacement measurements with [125I]-L-692,884 (▴).
The effect of 0.5 mg/kg (administered orally) of B-P2 (phenyl ester of B) and 0.3 mg/kg (administered orally) of B-P1 (ethyl ester of B) on ex vivo platelet aggregation in PRP upon stimulation with 20 μmol/L ADP (A) and platelet count in whole blood (B) in rhesus monkey 94-R021. The effect of 5 μg/kg IV bolus of A2-L and 20 μg/kg IV bolus of B on ex vivo platelet aggregation in PRP upon stimulation with 20 μmol/L ADP and platelet count in whole blood (C) and number of receptors occupied by B (D) in rhesus monkey 94-R021. (A and B) Inhibition of platelet aggregation by B-P1 (▾) or B-P2 (▴) as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. Platelet count measured in whole blood of rhesus monkey 94-R021 using an automated hematology analyzer (Biochem Immunosystems) upon dosing B-P1 (•) and B-P2 (▪). (C and D) Platelet count measured in whole blood of rhesus monkey 94-R021 using an automated hematology analyzer (Biochem Immunosystems) upon dosing A2-L (•) and B (▪). Inhibition of platelet aggregation by B (▾) or A2-L (⧫) as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. The number of GP IIb/IIIa receptors occupied by B as measured by competition displacement measurements with [125I]-L-692,884 (▴).

The effect of 10 μg/kg IV bolus of D, 10 μg/kg IV bolus of B, and 10 μg/kg IV bolus of B administered 30 minutes after 10 μg/kg IV bolus of D on ex vivo platelet aggregation in PRP stimulated with 20 μmol/L ADP (A) and platelet count in whole blood (B) in rhesus monkey 94-R021. Inhibition of platelet aggregation by B (▾) or D (▴) and sequential dosing of these compounds (○) as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. Platelet count measured in whole blood of rhesus monkey 94-R021 upon dosing D (•), B (▪), and B 30 minutes after D (◊).
The effect of 10 μg/kg IV bolus of D, 10 μg/kg IV bolus of B, and 10 μg/kg IV bolus of B administered 30 minutes after 10 μg/kg IV bolus of D on ex vivo platelet aggregation in PRP stimulated with 20 μmol/L ADP (A) and platelet count in whole blood (B) in rhesus monkey 94-R021. Inhibition of platelet aggregation by B (▾) or D (▴) and sequential dosing of these compounds (○) as measured by a change in light transmittance (PPP 100%) under stirring conditions (1,100 rpm) at 37°C in Biodata Platelet Aggregation Profiler. Platelet count measured in whole blood of rhesus monkey 94-R021 upon dosing D (•), B (▪), and B 30 minutes after D (◊).
Figure 8 shows the results of flow cytometric measurements of DDAbs in rhesus monkey and chimpanzee plasma. DDAbs were identified in rhesus monkey 94-R021 plasma only in the presence of B, using both rhesus monkey and human platelets. The FRA A2-L, which induced thrombocytopenia in chimpanzee A-264, had no effect on antibody binding in rhesus plasma. Similar results (data not shown) were obtained with 10 other FRAs, including A1-L, which induced thrombocytopenia in chimpanzee A-264. Figure 8 also demonstrates that B had no effect on binding of DDAbs from chimpanzee A-264 plasma. No DDAbs were detected in plasma of a control chimpanzee and rhesus monkey. When platelets were incubated with plasma samples of rhesus 94-R021 pretreated with 10 mmol/L EDTA for 1 hour at 37°C, the reaction of DDAbs with platelets was abolished (data not shown), as described above for chimpanzee A-264 plasma (Fig 3C).

Flow cytometric measurement of DDAbs in chimpanzee A-264, control chimpanzee, rhesus monkey 94-R021, and control rhesus monkey plasma alternatively using rhesus 94-R021 and human platelets in the presence or absence of 1 μmol/L of B or A2-L. Data represent the mean ± SD from three measurements. Binding of DDAbs is plotted as a relative fluorescence of FITC-labeled secondary antibody.
Flow cytometric measurement of DDAbs in chimpanzee A-264, control chimpanzee, rhesus monkey 94-R021, and control rhesus monkey plasma alternatively using rhesus 94-R021 and human platelets in the presence or absence of 1 μmol/L of B or A2-L. Data represent the mean ± SD from three measurements. Binding of DDAbs is plotted as a relative fluorescence of FITC-labeled secondary antibody.
Reaction of DDAbs from chimpanzee A-264 and rhesus 94-R021 plasma with platelets from type I Glanzmann’s thrombasthenia patient.
As shown in Table 3, DDAbs from chimpanzee A-264 as well as rhesus monkey 94-R021 plasma failed to react with platelets from a patient with type I Glanzmann’s thrombasthenia. This further supports the critical role of GP IIb/IIIa in binding of DDAbs from chimpanzee A-264 and rhesus 94-R021 plasma to platelets.
Reaction of DDAbs From Chimpanzee A-264, Rhesus Monkey 94-R021, and Control Chimpanzee Plasma With Platelets From a Glanzmann’s Thrombasthenic Patient and a Control Human Subject
| Platelets | Plasma | ||
|---|---|---|---|
| Chimpanzee A-264 | Rhesus 94-R021 | Control Chimpanzee | |
| Glanzmann’s patient | |||
| A1-L | 1.2 | 1.0 | 1.0 |
| A2-L | 1.1 | 1.1 | 1.0 |
| B | 1.1 | 1.2 | 1.1 |
| Control human subject | |||
| A1-L | 2.3 | 1.1 | 1.1 |
| A2-L | 5.2 | 1.0 | 1.2 |
| B | 1.2 | 3.3 | 1.0 |
| Platelets | Plasma | ||
|---|---|---|---|
| Chimpanzee A-264 | Rhesus 94-R021 | Control Chimpanzee | |
| Glanzmann’s patient | |||
| A1-L | 1.2 | 1.0 | 1.0 |
| A2-L | 1.1 | 1.1 | 1.0 |
| B | 1.1 | 1.2 | 1.1 |
| Control human subject | |||
| A1-L | 2.3 | 1.1 | 1.1 |
| A2-L | 5.2 | 1.0 | 1.2 |
| B | 1.2 | 3.3 | 1.0 |
The reaction of DDAbs expressed as relative fluorescence (fluorescence in the presence of the drug/fluorescence in the absence of the drug) obtained in the flow cytometric assay (for details, see Materials and Methods). Values ≥2 are considered a positive indication of DDAb binding. Values of relative fluorescence represent an average of two independent measurements.
Detection of DDAbs using purified GP IIb/IIIa coated on ELISA microplates.
DDAbs from chimpanzee A-264 plasma reacted in the presence of A2-L with purified GP IIb/IIIa coated on ELISA microplates in the presence of A2-L, providing a ratio of 7.4 (average of 2 independent measurements) for absorbance in the presence over the absorbance in the absence of A2-L. DDAbs failed to react with purified GP IIb/IIIa coated in the absence of A2-L. This indicates that the epitopes recognized by DDAbs are exposed on the surface of coated GP IIb/IIIa only in the presence of the drug.
Effect of rabbit antihuman GP IIb/IIIa polyclonal antibody (RPAb).
RPAb raised against purified human GP IIb/IIIa blocks platelet aggregation stimulated by ADP and the binding of fibrinogen and the monoclonal antibody PAC1 to activated platelets.35 RPAb abolished binding of DDAbs from both chimpanzee A-264 and rhesus 94-R021 plasma to all primate platelets tested (data not shown), thus indicating that the DDAbs from chimpanzee and rhesus monkey platelets recognize epitopes on primate GP IIb/IIIa.
Screening for preexisting DDAbs in human plasma samples.
The discovery of pre-existing DDAbs in plasma samples of chimpanzee A-264 and rhesus monkey 94-R021 and the consequent drug-induced thrombocytopenia in these primates stimulated a screening effort to determine the incidence of such DDAbs in humans. We screened 1,032 human plasma samples from healthy, anonymous blood donors using the flow cytometric assay described earlier and the FRAs that induced thrombocytopenia in chimpanzee A-264. Results are summarized in Table4. This screening identified 11 (1.1% ± 0.6%) plasma samples positive for A2-L–dependent and 8 (0.8% ± 0.6%) samples with A1-L–dependent antibodies. Only four positive plasma samples were positive for DDAbs using both FRAs (Table4). All positive plasma samples were then tested using other FRAs. As shown in Table 4, B and A3 stimulated binding of DDAbs to human platelets in some of the positive plasma samples. The flow cytomeric method could underestimate the incidence of DDAbs in human plasma if low-affinity DDAbs are lost during the wash step.
Cross Reactivity of Positive Human Plasma Samples for DDAbs
| Sample No. | A2-L | A1-L | A3 | B |
|---|---|---|---|---|
| 7 | + | + | + | + |
| 46 | + | + | + | + |
| 69 | + | − | + | + |
| 116 | + | − | − | − |
| 138 | − | + | − | − |
| 145 | − | + | − | − |
| 258 | − | + | − | − |
| 260 | − | + | − | − |
| 366 | + | − | − | − |
| 407 | + | − | − | − |
| 460 | + | + | − | − |
| 471 | + | − | + | − |
| 883 | + | − | + | − |
| 931 | + | − | − | − |
| 951 | − | − | + | − |
| 952 | − | − | + | − |
| 1051 | + | + | + | + |
| 1084 | − | − | − | + |
| Sample No. | A2-L | A1-L | A3 | B |
|---|---|---|---|---|
| 7 | + | + | + | + |
| 46 | + | + | + | + |
| 69 | + | − | + | + |
| 116 | + | − | − | − |
| 138 | − | + | − | − |
| 145 | − | + | − | − |
| 258 | − | + | − | − |
| 260 | − | + | − | − |
| 366 | + | − | − | − |
| 407 | + | − | − | − |
| 460 | + | + | − | − |
| 471 | + | − | + | − |
| 883 | + | − | + | − |
| 931 | + | − | − | − |
| 951 | − | − | + | − |
| 952 | − | − | + | − |
| 1051 | + | + | + | + |
| 1084 | − | − | − | + |
Screening of 1,032 human plasma samples for DDAbs using flow cytometric assay (for details, see Materials and Methods). The samples shown above were positive to at least one of the listed antagonists.
DISCUSSION
These studies document the sudden appearance of profound thrombocytopenia in one chimpanzee and one rhesus monkey upon administration of the highly GP IIb/IIIa selective, potent FRAs A1-L, A2-L, or B at doses that completely inhibited platelet aggregation. Flow cytometric analysis identified DDAbs in the plasma of both primates. The antibodies from these animals reacted with platelets from all primates tested only in the presence of the compound that induced thrombocytopenia in that particular animal. Although these two cases of thrombocytopenia are similar in the precipitous decrease in platelet count immediately after drug administration, there is an obvious difference in the rate of platelet recovery.
Kinetic studies of platelets in dogs with profound thrombocytopenia induced by the infusion of an antiglycoprotein IIb/IIIa monoclonal antibody demonstrated a dose-dependent precipitous decrease in platelet count after administration of antibody.36 The recovery of platelet count was also dose-related, with 3 hours required after 200 μg/kg and 12 hours after 300 μg/kg of antibody.32 The onset of drug-induced thrombocytopenia in both primates described in this report immediately followed the dosing of the drug.
The DDAbs identified in plasma of both primates were found in samples collected before the dosing of the drug as well as in samples collected several months after the drug-induced thrombocytopenia. These findings suggest that the drug-dependent antiplatelet antibodies present in the plasma of both primates before the first exposure to FRAs could have mediated the observed thrombocytopenia.
FRAs A1-L and A2-L have high affinity for GP IIb/IIIa, and at the concentrations used in chimpanzee A-264, they had an identical effect on ex vivo platelet aggregation. Both compounds induced profound thrombocytopenia in chimpanzee A-264; however, flow cytometric measurements of DDAbs indicate that the amount of antibody bound to the platelets was lower when A1-L was used. Repeat incubation of platelets with chimpanzee plasma in the presence of A2-L increased the amount of bound antibody, whereas repeat incubation in the presence of A1-L had little effect. The titer of DDAbs is low; however, the affinity of the antibodies for the receptor must be high, because they survived the washing step in the flow cytometric procedure. Thus, one can speculate that the concentration and also the dissociation constant of A2-L–dependent antibodies in chimpanzee A-264 plasma should be much lower than the concentration of GP IIb/IIIa receptors. Platelets were incubated with plasma at concentrations of 0.5 to 1.0 × 108 platelets/mL, which, assuming that the number of receptors per platelet is 57,000 ± 12,000 (see Table 1), gives a receptor concentration of approximately 4 to 10 nmol/L. The estimated dissociation constants of A2-L–dependent antibodies should then be subnanomolar, and the concentration of such antibodies in chimpanzee plasma would be in the range of several nanomolar. The lower amount of platelet-bound A1-L–dependent antibodies found by flow cytometry likely indicates higher dissociation constants of these antibodies, which prevented titration of the receptors after repeat incubations of platelets with chimpanzee plasma. The cause of these differences in binding affinities of DDAbs may stem from different effects of A1-L and A2-L on the conformation of the receptor and thus variability in the exposed neoepitopes. We have recently demonstrated subtle differences in the exposure of neoepitopes on GP IIb/IIIa upon ligand binding as measured by monoclonal antibodies.37 The lack of binding of DDAbs from chimpanzee A-264 plasma in the presence of A1-R, the R-isomer of A1-L, highlights the sensitivity of neoantigen exposure to minor changes in the structure of FRAs. The fact that neither of the drugs that induced thrombocytopenia in chimpanzee A-264 affected the platelet count or induced binding of DDAbs in rhesus 94-R021 also underscores the importance of specific drug structure on the exposure of LIBS for specific pre-existing antibodies. The specificity of neoantigen exposure on the GP IIb/IIIa receptor to the ligand structure is likely an explanation for different DDAbs found with different drugs in human plasma samples. Similar effects of subtle changes in the structure of drugs on neoepitopes exposure were described previously for sulfonamide-dependent15 and quinine-dependent antibodies.38 An analysis of plasma samples from patients who experienced thrombocytopenia during clinical trials with FRAs would help to determine if the DDAbs are always implicated in drug-induced thrombocytopenia in humans.
Structural differences, and thus likely differences in the composition of the epitope between A3 and the compounds that induced thrombocytopenia, could explain why this compound had no effect on platelet count in either chimpanzee A-264 or rhesus monkey 94-R021. One could also speculate that the affinity of the drug for the receptor could have an important role, because both compounds that induced thrombocytopenia in chimpanzee A-264 have high affinity for GP IIb/IIIa with a half time for dissociation from the receptor of 20 and 45 minutes for A1-L and A2-L, respectively. The FRA B, which induced thrombocytopenia in rhesus 94-R021, has a lower affinity for the receptor than A1-L and A2-L, and the half time for dissociation from the receptor is also much shorter.29 Interestingly, the recovery of the platelet count after the administration of B to rhesus 94-R021 was much faster than it was in both cases of thrombocytopenia in chimpanzee A-264, with higher affinity compounds. The affinity of A3 for resting platelets is low29 (Kd, 640 nmol/L). Because receptor occupancy by the drug is required for binding of DDAbs to platelets (as demonstrated in this report with A2-L–dependent antibodies), the kinetics of drug binding that control the kinetics of antibody binding may be critical. This is supported by the correlation between the time course of thrombocytopenia and the rate of the clearance of the FRAs that induced thrombocytopenia in chimpanzee A-264 and rhesus monkey 94-R021. In other words, the platelet count recovers in the chimpanzee after several days, whereas in the rhesus monkey, it recovers after several hours.
It is generally understood that in immune thrombocytopenia the platelet-antiplatelet antibody complex is trapped via the Fcγ receptors in the reticuloendothelial system of the liver and spleen.12 36 Based on our results, we hypothesize that the mechanism by which the GP IIb/IIIa antagonists induce the antibody binding to platelets requires binding of the antagonists to the receptors. The binding to the receptors results in the conformational change and an exposure of new epitopes on the surface of the GP IIIb/IIIa. Antibodies in the plasma that bind to platelets only in the presence of the drug recognize these neo-epitopes, as demonstrated by flow cytometric measurements. Platelet-drug-antibody complex is then captured via the Fcγ receptors in the reticuloendothelial systems of the liver and the spleen. The removal of the antibodies and return of the platelets to the circulation or eventual destruction of sequestered platelets is likely controlled by the off-rate of antibodies, which depends on the off-rate and Kd of the drug.
Highly integrin GP IIb/IIIa-selective FRAs, which induced thrombocytopenia in both primates, suggest a critical role of GP IIb/IIIa in the mechanism of thrombocytopenia. DDAb binding to primate platelets was abolished by incubating platelets either with EDTA at 37°C or with a rabbit antihuman GP IIb/IIIa polyclonal antibody that blocks platelet aggregation and binding of fibrinogen to platelets. These results, together with the identification of DDAbs using an ELISA and coated purified GP IIb/IIIa, confirmed that DDAbs bind to epitopes on GP IIb/IIIa. Flow cytometric measurements of DDAbs using platelets from a patient with type I Glanzmann’s thrombasthenia further confirmed that the GP IIb/IIIa receptor is the target of DDAbs found in chimpanzee A-264 and rhesus monkey 94-R021 plasma. Structural analysis of the epitopes on GP IIb/IIIa by flow cytometric and plasmon resonance measurements of competition between peptides derived from sequences of GP IIb and GPIIIa and DDAbs are in progress. It is interesting that autoantibodies to epitopes on GPIIb/IIIa have been reported in a low percentage of otherwise healthy subjects in EDTA-dependent thrombocytopenia.39
In summary, these results provide evidence to suggest that the described drug-induced thrombocytopenia in chimpanzee A-264 and rhesus monkey 94-R021 is caused by circulating IgG type DDAbs. In the presence of specific FRA(s), these antibodies react with GP IIb/IIIa on all primate platelets tested. The DDAbs were found in plasma samples collected before the first exposure of the primates to any FRA. We further showed that the incidence of DDAbs for two FRAs that induced thrombocytopenia in chimpanzee A-264 was approximately 1% in 1,032 human plasma samples. It remains to be seen whether the apparent correlation between the frequency of plasma samples positive for DDAbs and the incidence of thrombocytopenia in clinical trials is just coincidence, thus implicating alternate mechanisms in drug-induced thrombocytopenia, or if the thrombocytopenia is purely immune mediated.
ACKNOWLEDGMENT
The authors greatly appreciate the significant contributions of Benny C. Askew, Melissa S. Egbertson, and John H. Hutchinson for design and synthesis of the FRAs used in these experiments; Maria T. Stranieri, Gary R. Sitko, Joan D. Glass, and Raymond F. Stupienski for the careful performance of numerous experiments with conscious nonhuman primates; and Charles Chang and Lee Gaul for the screening of multiple human plasma samples for the presence of DDAbs. We also thank Daniel M. Bollag for critical reading of the manuscript.
B.B. and J.J.C. contributed equally to this report.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. section 1734 solely to indicate this fact.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal