

Abstract
X-linked sideroblastic anemia (XLSA) in four unrelated male probands was caused by missense mutations in the erythroid-specific 5-aminolevulinate synthase gene (ALAS2). All were new mutations: T647C, C1283T, G1395A, and C1406T predicting amino acid substitutions Y199H, R411C, R448Q, and R452C. All probands were clinically pyridoxine-responsive. The mutation Y199H was shown to be the first de novo XLSA mutation and occurred in a gamete of the proband’s maternal grandfather. There was a significantly higher frequency of coinheritance of the hereditary hemochromatosis (HH)HFE mutant allele C282Y in 18 unrelated XLSA hemizygotes than found in the normal population, indicating a role for coinheritance ofHFE alleles in the expression of this disorder. One proband (Y199H) with severe and early iron loading coinherited HH as a C282Y homozygote. The clinical and hematologic histories of two XLSA probands suggest that iron overload suppresses pyridoxine responsiveness. Notably, reversal of the iron overload in the Y199H proband by phlebotomy resulted in higher hemoglobin concentrations during pyridoxine supplementation. The proband with the R452C mutation was symptom-free on occasional phlebotomy and daily pyridoxine. These studies indicate the value of combined phlebotomy and pyridoxine supplementation in the management of XLSA probands in order to prevent a downward spiral of iron toxicity and refractory anemia.
THE SIDEROBLASTIC ANEMIAS are a heterogeneous group of disorders characterized by anemia of varying severity, hypochromic peripheral erythrocytes, progressive accumulation of iron, and the presence of ringed sideroblasts in the bone marrow.1 The disorder may be either inherited or acquired. X-linked sideroblastic anemia (XLSA; OMIM 301300)2 is the most common of the inherited forms of sideroblastic anemia and, with the discovery that this microcytic anemia is the result of mutations in the erythroid-specific isozyme of 5-aminolevulinate synthase,3 it is also the best understood at the molecular level.4-10 5-Aminolevulinic acid synthase [E.C. 2.3.1.37; ALAS] is the first and rate-limiting enzyme in heme biosynthesis, and the erythroid isozyme, ALAS2, is specifically expressed in erythroid tissues at high levels to provide heme for hemoglobin (Hb) synthesis. The X-chromosomal linkage of this hereditary sideroblastic anemia has been documented since 1946,11,12 and the human ALAS2 gene has been localized to the chromosomal region Xp11.21.13 Most patients with XLSA are, to some extent, responsive to pyridoxine,14 which is metabolized to pyridoxal 5′-phosphate (PLP), the cofactor for ALAS2. The defective activity of this enzyme in bone marrow erythroblasts in patients with XLSA15,16 diminishes heme biosynthesis, leading to insufficient protoporphyrin IX to use all of the available iron and therefore to reduced Hb concentrations and elevated tissue iron. This causes expansion of the erythroid marrow and ineffective erythropoiesis, resulting in increased iron absorption.17 18 The subsequent progressive toxic accumulation of iron occurs in most tissues and is particularly damaging to the liver, heart, pancreas, and pituitary. If untreated, the continuing iron deposition leads to arthritic signs, endocrine disorders (including delayed growth, impotence, and diabetes), cirrhosis of the liver, and heart failure.
Clinical management of uncomplicated XLSA involves attention paid to the anemia, the monitoring and depletion of iron stores, family studies to identify additional at-risk individuals, and genetic counseling. In the past, XLSA patients sometimes died in infancy due to severe anemia.19 However, with the advent of more accurate diagnosis and better clinical management, the cause of death is more often due to the toxic effects of the progressive iron overload resulting from sustained iron absorption and/or blood transfusions used to treat the anemia.1 Direct mutation analysis of patients with XLSA as described here enables diagnosis of heterozygotes, as well as correlation of the proband’s clinical state with specific mutations in the ALAS2 gene and thus an understanding of the likely prognosis and response to treatment of the anemia for other patients with these mutations.
Iron overload is also a feature of hereditary hemochromatosis (HH), an autosomal recessive disorder with an estimated gene frequency of 6% to 8% in the white population.20 Disease penetrance in HH homozygotes is low, and clinically significant iron overload generally presents as a function of age, diet, or exposure to alcohol.21 Several investigators have examined whether heterozygosity for the HH-associated human leukocyte antigen (HLA) haplotypes A3, B7, and B14 were associated with more severe iron overload in acquired idiopathic sideroblastic anemia and hereditary sideroblastic anemia patients.18,22-24 However, the results of these previous HLA haplotype studies were inconclusive, largely due to the small number of patients examined and genetic heterogeneity of HH. Recently a candidate gene, HFE, distal to the major histocompatibility locus region at 6p was identified for HH25 and a single mutation reported to account for the majority of HH patients in whites.25-27
In this report, we describe four different mutations of theALAS2 gene in unrelated patients with pyridoxine-responsive XLSA. Their clinical and genetic heterogeneity is documented and an increased frequency of the HFE mutant allele C282Y was found in XLSA hemizygotes. Pyridoxine responsiveness and/or Hb concentrations increased with iron depletion. XLSA heterozygotes were variably affected and highlight the importance of molecular diagnosis and appropriate therapy.
CASE REPORTS AND METHODS
Case report: Family 5.
Note that families 1 to 4 were previously published, as referenced in Table 3. The proband of family 5 (of Irish descent; born February 2, 1968) presented at 16 years of age with hepatosplenomegaly and severe microcytic (mean corpuscular volume [MCV], 60 fL; see Table1 for normal ranges), hypochromic (mean corpuscular hemoglobin [MCH], 18.9 pg) anemia (Hb, 9.3 g/dL) that was refractory to 6 to 7 months of oral iron supplementation. Subsequent liver function tests were abnormal and liver biopsy showed excess iron and precirrhotic changes. Serum ferritin levels were reported to be greater than 1,000 μg/L and transferrin was fully saturated. He was treated first with pyridoxine with no hematologic response and then with desferrioxamine, pyridoxine, and folic acid (5 mg/d) simultaneously for approximately 5 years (Fig1A). Hb increased to 12 g/dL, by which time the serum ferritin level had decreased to a low normal value. A second liver biopsy during this time showed only hemosiderin. The changes in Hb during this period were quite faithfully the inverse of the changes in serum ferritin concentration. He subsequently presented (off all treatment) at the University Hospital of Wales with Hb 9.6 g/dL, transferrin saturation 69%, and serum ferritin level 233 μg/L (month 100, Fig 1A). His bone marrow erythroblasts showed 20% ringed sideroblasts with a normal karyotype. Marrow iron turnover (MIT) was increased at 545 μmol/L blood/d (normal range, 70 to 140 μmol/L blood/d) with 87% ineffective erythropoiesis (normal range, 20% to 30%). The free erythrocyte protoporphyrin level was 0.7 μmol/L red blood cells (RBCs; normal range, 0.4 to 1.7 μmol/L RBCs). Subsequent pyridoxine therapy, while storage iron levels remained high, modestly raised Hb from approximately 7.5 g/dL to a plateau of 9.5 g/dL. Once sufficient iron was removed by phlebotomy to reduce serum ferritin to normal levels and to begin reducing transferrin saturation, the pyridoxine responsiveness become even more apparent, with further increases in Hb, MCV, and MCH (Fig 1A). Subsequent continued removal of iron resulted in a transient iron-deficiency anemia. With continued pyridoxine supplementation and low iron, the most recent Hb value was increased to 11.1 g/dL.
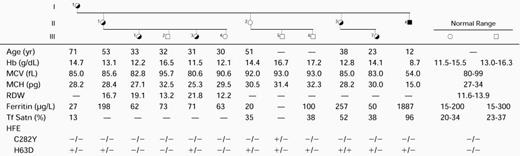
Hematologic Status of the Members of Family 6
 |
|
Abbreviations: RDW, red blood cell distribution width; Tf Satn, transferrin saturation.


Hematologic and iron status in response to pyridoxine supplementation and iron removal by desferrioxamine chelation and/or phlebotomy. The probands were reliable regarding pyridoxine self-administration and were in good health during the critical periods of pyridoxine administration (family 5: months 108-113 and 130-135; family 6: Months 140-145 and 160-165). Repeat blood counts were confirmatory of the observed trends. (A.) Family 5 (Y199H) proband. (B). Family 6 (R411C) proband.
Hematologic and iron status in response to pyridoxine supplementation and iron removal by desferrioxamine chelation and/or phlebotomy. The probands were reliable regarding pyridoxine self-administration and were in good health during the critical periods of pyridoxine administration (family 5: months 108-113 and 130-135; family 6: Months 140-145 and 160-165). Repeat blood counts were confirmatory of the observed trends. (A.) Family 5 (Y199H) proband. (B). Family 6 (R411C) proband.
There was no family history of anemia. Erythrocytes from the proband’s brother, father, maternal grandmother, and five maternal and three paternal aunts all had a normal, single distribution of cell size and Hb content. However, the mother, who had normal values for Hb and MCH, had an abnormal RBC distribution width (RDW) of 18.5 and showed a bimodal distribution of erythrocytes containing a minor population of microcytic/hypochromic cells. Serum ferritin and transferrin saturation levels were normal in all relatives, except for the maternal grandmother (83 years), who had a slightly elevated serum ferritin value (212 μg/L), but normal transferrin saturation (33%).
Case report: Family 6.
This family was previously shown to have XLSA by Holmes et al.28 The proband (II.5; born December 12, 1960) presented at age 8 with hypochromic (Hb, 7.5 g/dL), microcytic anemia and transferrin saturation of 20%. The clinical summary of his observations and therapy during the following 28 years is shown in Fig1B. Before initiation of 3 mg/d pyridoxine, he received 20 injections of iron with no effect on his anemia. Throughout his time on low-dose pyridoxine, he took oral iron and 5 mg/d folic acid. With 3 mg/d pyridoxine, Hb increased 11.2 g/dL over the first 2 years. However, by the end of 10 years, transferrin saturation increased to 95% and serum ferritin to 1,450 μg/L, while MCH and MCV decreased significantly (Fig 1B). By the time of his referral to the University Hospital of Wales (month 138), he had stopped all medication and bone marrow erythroblasts were 75% ringed sideroblasts and MIT was about six times normal (706 μmol/L blood/d) with 90% ineffective erythropoiesis. Without pyridoxine therapy, Hb decreased dramatically but subsequently rebounded in the first 2 months of oral pyridoxine and thiamine (200 mg/d), and then stabilized at approximately 9 g/dL. This was repeated with a smaller oscillation. For the next 10 years, pyridoxine was maintained at 200 mg/d, during which time Hb declined steadily to a new plateau of 8 g/dL as serum ferritin increased from 1,200 μg/L to 3,000 μg/L. At this time, gamma glutamyl transferase and AST were elevated, glucose intolerance had developed, and liver biopsy indicated iron overload and fatty changes. Treatment with subcutaneous desferrioxamine was begun. This caused an immediate improvement in liver function and the glucose intolerance stabilized. Over the following 30 months on desferrioxamine, the proband’s serum ferritin level decreased to normal values (227 μg/L), but transferrin saturation remained high (79%) and the patient developed clinical evidence of cardiomyopathy, congestive heart failure, and life-threatening cardiac arrhythmias. After cessation of desferrioxamine, serum ferritin began increasing (324 μg/L) and transferrin saturation was 86%, indicating that tissues were still iron-loaded. During this period, MCV and MCH remained low, except when transferrin saturation transiently decreased to approximately 50%.
Previously, the proband’s two sisters and three nieces had dimorphic erythrocyte populations, whereas his mother’s erythrocytes appeared normal.28 The more recent hematologic status of family 6, summarized in Table 1, showed no change in these findings. The completely normal RBC size distribution for the mother (Fig2D) is compared with the broad abnormal population of the proband and the dimorphic populations of his youngest sister and her daughter (Fig 2). Although the proband’s mother has developed late-onset diabetes, her hematologic iron status was normal, as it was in all relatives except for the proband’s sister (II.3) who had elevated iron (serum ferritin, 257 μg/L) without anemia.

Red blood cell (RBC) size distribution for family 6 hemizygous proband and heterozygotes. (—•—) Relative frequency of a particular RBC cell size (----); range of RBC size distributions of normal individuals. MCV is indicated for each individual. (A) Profile for family 6 proband II.4 (XLSA genotype and pedigree numbers are from Table 1). (B) Profile for the proband’s sister, II.3. (C) Profile for the proband’s niece, III.7. (D) Profile for proband’s mother, I.1.
Red blood cell (RBC) size distribution for family 6 hemizygous proband and heterozygotes. (—•—) Relative frequency of a particular RBC cell size (----); range of RBC size distributions of normal individuals. MCV is indicated for each individual. (A) Profile for family 6 proband II.4 (XLSA genotype and pedigree numbers are from Table 1). (B) Profile for the proband’s sister, II.3. (C) Profile for the proband’s niece, III.7. (D) Profile for proband’s mother, I.1.
Case report: Family 7.
The proband (born in 1974) presented with microcytic, hypochromic anemia (Hb, 7.0 g/dL; MCV 69 fL) at the age of 11. He was initially diagnosed with autoimmune hypothyroidism, which his mother and a cousin also have. X-ray showed a markedly porotic spine along with significant cardiac enlargement. He was treated with iron, folate supplements, and thyroxine replacement. After 2 months, Hb increased to 10.4 g/dL and MCV was 72 fL. He was examined again at the age of 17, when his Hb was 10.6 g/dL, MCV 69 fL, serum iron 29 μmol/L, total iron-binding capacity (TIBC) 66 μmol/L, transferrin saturation 44%, serum ferritin 48 μg/L, and B12 and folate levels normal. Bone marrow examination showed normal cellularity with dyserythropoiesis, while iron staining showed 29% ring sideroblasts. Bone marrow chromosome studies were normal. When readmitted for mutation analysis, the patient showed a modest response to high-dose pyridoxine (300 to 400 mg/d); initially, Hb increased from 9.5 to 11.9 g/dL, but gradually decreased to 10.3 g/dL over the next 2 years of this pyridoxine supplementation. Serum iron (SI) measurements on two occasions (29 μmol/L and 42 μmol/L) showed a tendency to increased transferrin saturation, but serum ferritin levels remained low. The proband’s sister was slightly anemic (Hb, 11.4 g/dL; MCV, 82 fL). The mother (Hb, 12 g/dL; MCV, 83 fL) had an RDW of 21.2 and occasional microcytic, hypochromic cells in her blood film. The maternal grandmother (Hb, 12.5 g/dL; MCV, 87 fL) had a normal RDW and blood films.
Case report: Family 8.
The proband, a 30-year-old man, was diagnosed in childhood with mild sideroblastic anemia with hypochromic, microcytic erythrocytes that was moderately responsive to pyridoxine (300 mg/d). Folic acid supplementation (1 mg/d) was also maintained. Iron overload had been avoided by four phlebotomies per year and the patient is in good health with continued phlebotomies and daily pyridoxine supplementation.
Molecular analysis of the erythroid ALAS2 gene.
Genomic DNA was isolated by standard techniques29 from peripheral blood or lymphoblastoid cell lines obtained after informed consent from the probands and other family members. Polymerase chain reaction (PCR) and sequence analysis of the ALAS2 gene were performed as previously described,3 4 using the oligonucleotides and annealing temperatures listed in Table2. The same primers were used for both PCR amplification and sequencing. The 5′ GC clamps and restriction sites in the primer sequences were originally included to facilitate subcloning, but currently, all sequencing is accomplished by direct sequencing of the amplified DNA. For confirmation of mutations by restriction analysis, the products from PCR amplification of exon 5 were digested with Sau3AI. Exon 9 was PCR-amplified with the alternative oligonucleotides 129 and 130 to give better restriction fragment size discrimination (Table 2) and PCR products were digested withHinP1I, BsrI, or BanII (New England Biolabs, Beverly, MA). All restriction digests were electrophoresed in 2% agarose (ultrapure grade; GIBCO BRL, Grand Island, NY) gels containing 0.1 μg/mL ethidium bromide.
PCR Primers and Conditions for Amplification ofALAS2 From Genomic DNA
| ALAS2 Region | Size (bp) | Temperature (°C) | Primer No. | Sense* | Oligonucleotides† |
|---|---|---|---|---|---|
| Promoter | 351 | 56 | 117 | + | 5′ GCCGCCAAGCTT‖AAAAAAGAAATTGCAAATCAATATGT 3′ |
| Region 1 | 313 | − | 5′ GCCGCCAAGCT‖TAACTTGTTGATAATTACCCAACTA 3′ | ||
| Promoter | 326 | 58 | 118 | + | 5′ GCCGCCAAGC‖TTCATAGGCGGGCTCTG 3′ |
| Region 2 | 314 | − | 5′ GCCGCCAA‖GCTTATGAGCTCAAACAGTCAGCTT 3′ | ||
| Promoter | 290 | 60 | 119 | + | 5′ GCCGCCAAGC‖TTACAACAACCGGGGATC 3′ |
| Region 3 | 316 | − | 5′ GCCGCCAAGCT‖TCTGGCTCTTCCCTATTT 3′ | ||
| Exon 1 | 323 | 60 | 136 | + | 5′ GCCGCCGAATTC‖CTAATTTTACTGTCCTATAGAG 3′ |
| 128 | − | 5′ GCCGCCGAATT‖CAGCTGGCAGACCAGAGATA 3′ | |||
| Exon 2 | 390 | 60 | 137 | + | 5′ GCCGCCGAATTC‖GAAGGGCAATAAGAGCA 3′ |
| 135 | − | 5′ GCCGCCGAATT‖CCCCAGGACCCTAACAT 3′ | |||
| Exon 3 | 242 | 58 | 158 | + | 5′ GCCGCCGAATT‖CATTAGATCTCAGCAATTAT 3′ |
| 159 | − | 5′ GCCGCCGAATTC‖GGTGGAACTTGACTCCA 3′ | |||
| Exon 4 | 334 | 56 | 143 | + | 5′ GCCGCCGAATTC‖AAACTTGAATTTTCATG 3′ |
| 144 | − | 5′ GCCGCCGAATTC‖GCCCTTCTGTACTGTTT 3′ | |||
| Exon 5 | 355 | 62 | 329 | + | 5′ ‖AGACTAGCCAGGGAGAGACT 3′ |
| 169 | − | 5′ GCCGCCGAATTC‖TTTCCATGTGTGGTTTTTC 3′ | |||
| Exon 6 | 334 | 58 | 145 | + | 5′ GCCGCCGAATT‖CTACCCAGTTCCTCGA 3′ |
| 146 | − | 5′ GCCGCCGAATTC‖GTAAACTGGATGCTGTAT 3′ | |||
| Exon 7 | 324 | 60 | 141 | + | 5′ GCCGCCGAA‖TTCTTTGCCAGGTCAAACC 3′ |
| 142 | − | 5′ GCCGCCGAATTC‖GACCAACACTAGTAAACAT 3′ | |||
| Exon 8 | 298 | 60 | 315 | + | 5′ GCCGCCG‖AATTCCACATTGGAGATGG 3′ |
| 151 | − | 5′ GCCGCCGAATT‖CCTCCTCTCTGGAGG 3′ | |||
| Exon 9 | 416 | 60 | 115 | + | 5′ GCCGCCGAATT‖CATGATCCTGTTGCTCT 3′ |
| 116 | − | 5′ GCCGCCGAATTC‖AGCGTGAGGCTCCCAGA 3′ | |||
| Exon 9 | 293 | 60 | 129 | + | 5′ GCCGCCGAATT‖CAGGCAAGGCCTTTGGCTGT 3′ |
| 130 | − | 5′ GCCGCCGAATTC‖CGGATGGGGATGATGTGGC 3′ | |||
| Exon 10 | 322 | 62 | 147 | + | 5′ GCCGCCGAATT‖CATCTGCTTAATGGAGCTA 3′ |
| 148 | − | 5′ GCCGCCGAATTC‖TCTCTTTCAGATCCTGGG 3′ | |||
| Exon 11 | 319 | 60 | 152 | + | 5′ GCCGCCGAATTC‖TGGAAGATCTAGTCTAAC 3′ |
| 153 | − | 5′ GCCGCCGAATT‖CACAACAAAGCAGAAGAC 3′ | |||
| 3′ end | 372 | 60 | 318 | + | 5′ GCCGCCGA‖ATTCACACCCCACCTGC 3′ |
| 319 | − | 5′ GCCGCCGAAT‖TCAATCCCTGGATTTTTATTG 3′ |
| ALAS2 Region | Size (bp) | Temperature (°C) | Primer No. | Sense* | Oligonucleotides† |
|---|---|---|---|---|---|
| Promoter | 351 | 56 | 117 | + | 5′ GCCGCCAAGCTT‖AAAAAAGAAATTGCAAATCAATATGT 3′ |
| Region 1 | 313 | − | 5′ GCCGCCAAGCT‖TAACTTGTTGATAATTACCCAACTA 3′ | ||
| Promoter | 326 | 58 | 118 | + | 5′ GCCGCCAAGC‖TTCATAGGCGGGCTCTG 3′ |
| Region 2 | 314 | − | 5′ GCCGCCAA‖GCTTATGAGCTCAAACAGTCAGCTT 3′ | ||
| Promoter | 290 | 60 | 119 | + | 5′ GCCGCCAAGC‖TTACAACAACCGGGGATC 3′ |
| Region 3 | 316 | − | 5′ GCCGCCAAGCT‖TCTGGCTCTTCCCTATTT 3′ | ||
| Exon 1 | 323 | 60 | 136 | + | 5′ GCCGCCGAATTC‖CTAATTTTACTGTCCTATAGAG 3′ |
| 128 | − | 5′ GCCGCCGAATT‖CAGCTGGCAGACCAGAGATA 3′ | |||
| Exon 2 | 390 | 60 | 137 | + | 5′ GCCGCCGAATTC‖GAAGGGCAATAAGAGCA 3′ |
| 135 | − | 5′ GCCGCCGAATT‖CCCCAGGACCCTAACAT 3′ | |||
| Exon 3 | 242 | 58 | 158 | + | 5′ GCCGCCGAATT‖CATTAGATCTCAGCAATTAT 3′ |
| 159 | − | 5′ GCCGCCGAATTC‖GGTGGAACTTGACTCCA 3′ | |||
| Exon 4 | 334 | 56 | 143 | + | 5′ GCCGCCGAATTC‖AAACTTGAATTTTCATG 3′ |
| 144 | − | 5′ GCCGCCGAATTC‖GCCCTTCTGTACTGTTT 3′ | |||
| Exon 5 | 355 | 62 | 329 | + | 5′ ‖AGACTAGCCAGGGAGAGACT 3′ |
| 169 | − | 5′ GCCGCCGAATTC‖TTTCCATGTGTGGTTTTTC 3′ | |||
| Exon 6 | 334 | 58 | 145 | + | 5′ GCCGCCGAATT‖CTACCCAGTTCCTCGA 3′ |
| 146 | − | 5′ GCCGCCGAATTC‖GTAAACTGGATGCTGTAT 3′ | |||
| Exon 7 | 324 | 60 | 141 | + | 5′ GCCGCCGAA‖TTCTTTGCCAGGTCAAACC 3′ |
| 142 | − | 5′ GCCGCCGAATTC‖GACCAACACTAGTAAACAT 3′ | |||
| Exon 8 | 298 | 60 | 315 | + | 5′ GCCGCCG‖AATTCCACATTGGAGATGG 3′ |
| 151 | − | 5′ GCCGCCGAATT‖CCTCCTCTCTGGAGG 3′ | |||
| Exon 9 | 416 | 60 | 115 | + | 5′ GCCGCCGAATT‖CATGATCCTGTTGCTCT 3′ |
| 116 | − | 5′ GCCGCCGAATTC‖AGCGTGAGGCTCCCAGA 3′ | |||
| Exon 9 | 293 | 60 | 129 | + | 5′ GCCGCCGAATT‖CAGGCAAGGCCTTTGGCTGT 3′ |
| 130 | − | 5′ GCCGCCGAATTC‖CGGATGGGGATGATGTGGC 3′ | |||
| Exon 10 | 322 | 62 | 147 | + | 5′ GCCGCCGAATT‖CATCTGCTTAATGGAGCTA 3′ |
| 148 | − | 5′ GCCGCCGAATTC‖TCTCTTTCAGATCCTGGG 3′ | |||
| Exon 11 | 319 | 60 | 152 | + | 5′ GCCGCCGAATTC‖TGGAAGATCTAGTCTAAC 3′ |
| 153 | − | 5′ GCCGCCGAATT‖CACAACAAAGCAGAAGAC 3′ | |||
| 3′ end | 372 | 60 | 318 | + | 5′ GCCGCCGA‖ATTCACACCCCACCTGC 3′ |
| 319 | − | 5′ GCCGCCGAAT‖TCAATCCCTGGATTTTTATTG 3′ |
(+) Sense primer; (−) antisense primer.
Vertical bar indicates the beginning of the ALAS2 genomic sequence. The annealing temperatures were set for the regions to the right of the vertical bar.
Polymorphism analysis of the ALAS2 intron 7 dinucleotide repeat.
The highly informative polymorphic ALAS2 intron 7 dinucleotide repeat was PCR-amplified from XLSA family 5 using 500 ng genomic DNA, 50 μmol/L of each dNTP, 1.5 mmol/L MgCl2, and 0.1 μmol/L each oligonucleotide in a 100-μL reaction volume using the conditions and primer sequences of Cox et al.30 Alleles were separated by electrophoresis in 4% MetaPhor agarose (FMC Bioproducts, Rockland, ME) containing 0.1 μg/mL ethidium bromide. Molecular weight was estimated from a semilog plot comparing the mobility of the 100-bp ladder standard with the ALAS2 intron 7 CA repeat alleles.
Molecular analysis of the HFE gene.
The HFE mutations, C282Y and H63D, were detected as described using the 20-mer PCR primers,25 with the following modifications. The HFE gene was PCR-amplified using 1 μg genomic DNA, 50 μmol/L of each dNTP, 1.5 mmol/L MgCl2, and 1 μmol/L of each oligonucleotide in a 100-μL reaction volume.25 The annealing temperature was 60°C. The PCR products containing the C282 codon were restricted with RsaI (New England Biolabs); the C282Y allele was distinguished from the normal allele as the mutation introduced an additional RsaI restriction site. The H63D mutation resulted in the creation of aSau3AI site.
Clinical diagnostic methods.
Routine hematologic measurements at the University Hospital of Wales for families 5 and 6 were obtained using Bayer Technicon (Tarytown, NY) models H6000, H1, H2, and H3 automatic cell analyzers. Data for analysis of erythrocyte size distribution used in Fig 2 was obtained on a Coulter Counter model S-Plus IV (Coulter Electronics, Hialeah, FL) as described and plotted using interpolation instead of curve-fitting.31 Serum ferritin levels for families 5, 6, and 8 were measured by enzyme-linked immunosorbent assay (ELISA)32; SI and TIBC were measured using a chromogenic assay,33 and erythrocyte protoporphyrin was measured fluorimetrically.34 Ferrokinetic analysis was performed by the method of Cavill et al.35
RESULTS
Identification of missense mutations of the ALAS2 gene in XLSA patients.
Genomic DNA was isolated from four families with pyridoxine-responsive XLSA. Each exon of the ALAS2 gene, including 50 to 150 nt of flanking intron sequence, 1 kb of 5′, and 350 nt of 3′ flanking sequence, was PCR-amplified and sequenced.
A single point mutation was found in exon 5 of the ALAS2 gene from the proband of family 5. This T to C transversion at nt 647 predicted the substitution of histidine for tyrosine at residue 199 (Y199H). This mutation created a Sau3AI restriction site in exon 5. Restriction of PCR-amplified ALAS2 exon 5 from the proband resulted in fragments of 120, 117, 99, and 19 bp, while the PCR products from normal control individuals yielded fragments of 237, 99, and 19 bp, (Fig 3A). PCR and restriction analysis showed that the proband’s mother was a heterozygote for the Y199H mutation, while all other family members were normal (Fig 3A).

Restriction analysis for confirmation of the exon 5 mutation, analysis of mutation origin, and genotype analysis for the C282Y HFE mutation in family 5. (A) Sau3AI restriction of exon 5 PCR products. (B) Polymorphic allele haplotype for the CA repeat in intron 7 of the ALAS2 gene. (C) RsaI restriction analysis of PCR products encompassing the C282 codon of theHFE gene.
Restriction analysis for confirmation of the exon 5 mutation, analysis of mutation origin, and genotype analysis for the C282Y HFE mutation in family 5. (A) Sau3AI restriction of exon 5 PCR products. (B) Polymorphic allele haplotype for the CA repeat in intron 7 of the ALAS2 gene. (C) RsaI restriction analysis of PCR products encompassing the C282 codon of theHFE gene.
Sequence analysis of genomic DNA from the proband in family 6 identified a C to T transition at nt 1283, which predicted the substitution of cysteine for arginine at residue 411 (R411C). This mutation eliminated a HinP1I site in exon 9 and restriction analysis of PCR-amplified ALAS2 exon 9 was used to determine the carrier status of the female members of this family (Fig4A). With one exception, these results correlated with the carrier assignments made by Holmes et al28 based on erythrocyte morphology. Digestion of the 293-bp PCR product from the proband resulted in fragments of 199, 76, and 18 bp, compared with fragments of 123, 76, 76, and 18 bp in PCR products from normal individuals. Restriction analysis identified as heterozygotes the proband’s mother (I.1), two of his sisters (II.1 and II.3) and three of his nieces (III.1, III.3, and III.7) (Fig 4A). The remaining family members were normal. Of note, the mother of the proband, although an obligate heterozygote confirmed at the DNA level, had a completely normal erythrocyte profile (Fig 2D), presumably resulting from skewed lyonization favoring the normal ALAS2allele.

Restriction analysis for confirmation of the exon 9 mutations. In all gels, the Std. lane contained the size standards generated by HaeIII digestion of ▹X174, (Pharmacia, Piscataway, NJ). (A) HinP1I restriction of exon 9 PCR products from the family 6 proband and other family members with the R411C mutation. (B) Elimination of a BanII restriction site confirms the presence of the R448Q mutation in members of family 7. (C)BsrI restriction of the PCR product from exon 9 of the family 9 proband (lane 3) confirmed the presence of the R452C mutation. The 2 normal controls (lanes 2 and 4) remained uncut.
Restriction analysis for confirmation of the exon 9 mutations. In all gels, the Std. lane contained the size standards generated by HaeIII digestion of ▹X174, (Pharmacia, Piscataway, NJ). (A) HinP1I restriction of exon 9 PCR products from the family 6 proband and other family members with the R411C mutation. (B) Elimination of a BanII restriction site confirms the presence of the R448Q mutation in members of family 7. (C)BsrI restriction of the PCR product from exon 9 of the family 9 proband (lane 3) confirmed the presence of the R452C mutation. The 2 normal controls (lanes 2 and 4) remained uncut.
A missense mutation, a G to A transition, was identified in the proband of family 7 at nt 1395, predicting the substitution of glutamine for arginine at residue 448 (R448Q). This mutation eliminated aBanII restriction site in exon 9, and restriction of PCR-amplified ALAS2 exon 9 from the proband confirmed the mutation and identified carrier females in the family (Fig 4B). Restriction of the 293-bp PCR product from the proband resulted in fragments of 154 and 139 bp, whereas fragments of 139, 100, and 54 bp were observed from PCR products of normal individuals. The proband’s sister, mother, and grandmother were identified as heterozygotes for the R448Q mutation (Fig 4B). An aunt and two male cousins were normal by restriction analysis with BanII (data not shown).
In family 8, a C to T transition was identified at nt 1406, predicting the substitution of cysteine for arginine at residue 452 (R452C). The mutation introduced a BsrI restriction site in exon 9 and restriction analysis of PCR-amplified ALAS2 exon 9 from the proband confirmed the mutation; BsrI digestion of the 293-bp PCR product from the proband resulted in fragments of 196 and 97 bp, whereas PCR products from the normal controls remained uncut (Fig 4C). We have recently identified this mutation in another unrelated XLSA family (D.F. Bishop, unpublished data, July 1998), confirming it is the causative mutation.
The Y199H, R411C, R448Q, and R452C mutations were not found in any of 100 normal alleles examined by PCR and restriction analysis withSau3AI, HinP1I, BanII, and BsrI, respectively, in unrelated whites (data not shown), indicating that none of the four mutations was a polymorphism. In all cases, the missense mutations described were the only change identified in the 11 exons, the intron-exon junctions, and the 5′ and 3′ flanking sequences of genomic DNA from the four probands.
Homology comparison.
Homology comparisons with 18 other ALAS sequences demonstrated that the mutations occurred in regions that were highly conserved, particularly among higher organisms (not shown). The tyrosine residue at 199 (family 5) was invariant among all ALAS sequences, as was the family 6 mutation: the arginine substitution by cysteine at residue 411. The arginine residues in the two families with mutations R448Q and R452C (families 7 and 8, respectively) were both conserved in higher organisms, but not in unicellular organisms.
Parental origin of a de novo ALAS2 mutation.
Since none of proband 5’s maternal aunts or maternal grandmother were heterozygous for the Y199H mutation, the mutation must have been expressed de novo in the proband’s mother (Fig 3A). To determine the parental origin of the de novo mutation, the ALAS2 intron 7 CA dinucleotide repeat30 was PCR-amplified from members of XLSA family 5 (Fig 3B). The proband was hemizygous for an allele with a mobility consistent with that of the A5 allele, and his mother (II.3), three of his maternal aunts (II.4, II.5, and II.7), and his grandmother (I.1) were heterozygous for the A1 and A5 alleles. The proband’s maternal grandfather must have been hemizygous for the A5 allele, as two of his daughters were A5 homozygotes (II.6 and II.8). Thus, the A1 allele in the proband’s mother was inherited from the grandmother and the A5 allele associated with the XLSA mutation was inherited from the grandfather. The mutation must have arisen de novo on a grandpaternal A5 gamete, as none of his other daughters, who are obligate heterozygotes for the paternal A5 allele, were carriers of the Y199H mutation.
Identification of HH gene mutations in XLSA patients.
The probands and family members from the four XLSA pedigrees described here and the four families previously reported3,4 6 were screened for the HH gene (HFE) mutations C282Y and H63D by PCR amplification of genomic DNA and restriction analysis of PCR products as described in the Methods. The proband of family 5 (Y199H) was homozygous for the HFE C282Y (Fig 3C) mutation and normal for H63D (not shown). His parents, his brother, two maternal aunts, and one paternal aunt were heterozygous for the C282Y mutation and normal for H63D, while three maternal aunts were C282Y normal and heterozygous for H63D. The maternal grandmother was a compound heterozygote for C282Y and H63D. The coinheritance of the ALAS2 mutation Y199H and homozygosity for the HFE mutation C282Y resulted in large accumulations of iron. Even after earlier chelation therapy, approximately 5.8 g of iron was subsequently removed by phlebotomy before storage was normalized.
The probands of families 3 and 6 were heterozygous for HFEmutation H63D, while the probands of families 1, 2, 4, 7, and 8 were normal (Table 3). A sister (II.3) of the proband of family 6 was homozygous for HFE H63D (Table 1). Aside from the XLSA probands, elevated iron stores were found only in the HFE H63D homozygote (Table 1) and the C282Y/H63D compound heterozygote (serum ferritin, 212 μg/L).
HFE Genotype of XLSA Probands
| Family No. | Reference | ALAS2 Mutation | ALAS2 Allele | No. of Probands | C282Y Genotype | H63D Genotype |
|---|---|---|---|---|---|---|
| 1 | 3 | I476N | Hemi | 1 | −/− | −/− |
| 2 | 4 | F165L | Hemi | 1 | −/− | −/− |
| 3 | 6 | K299Q | Hemi | 1 | −/− | +/− |
| 4 | 6 | A172T | Het | 1 | −/− | −/− |
| 5 | 3-150 | Y199H | Hemi | 1 | +/+ | −/− |
| 6 | 3-150 | R411C | Hemi | 1 | −/− | +/− |
| 7 | 3-150 | R448Q | Hemi | 1 | −/− | −/− |
| 8 | 3-150 | R452C | Hemi | 1 | −/− | −/− |
| — | 36 | — | Hemi | 1 | +/− | +/− |
| — | 3-151 | — | Hemi | 1 | +/− | +/− |
| — | 3-151 | — | Hemi | 1 | +/− | −/− |
| — | 3-151 | — | Hemi | 3 | −/− | +/− |
| — | 3-151 | — | Hemi | 6 | −/− | −/− |
| — | 3-151 | — | Het | 1 | −/− | +/− |
| — | 3-151 | — | Het | 2 | −/− | −/− |
| Family No. | Reference | ALAS2 Mutation | ALAS2 Allele | No. of Probands | C282Y Genotype | H63D Genotype |
|---|---|---|---|---|---|---|
| 1 | 3 | I476N | Hemi | 1 | −/− | −/− |
| 2 | 4 | F165L | Hemi | 1 | −/− | −/− |
| 3 | 6 | K299Q | Hemi | 1 | −/− | +/− |
| 4 | 6 | A172T | Het | 1 | −/− | −/− |
| 5 | 3-150 | Y199H | Hemi | 1 | +/+ | −/− |
| 6 | 3-150 | R411C | Hemi | 1 | −/− | +/− |
| 7 | 3-150 | R448Q | Hemi | 1 | −/− | −/− |
| 8 | 3-150 | R452C | Hemi | 1 | −/− | −/− |
| — | 36 | — | Hemi | 1 | +/− | +/− |
| — | 3-151 | — | Hemi | 1 | +/− | +/− |
| — | 3-151 | — | Hemi | 1 | +/− | −/− |
| — | 3-151 | — | Hemi | 3 | −/− | +/− |
| — | 3-151 | — | Hemi | 6 | −/− | −/− |
| — | 3-151 | — | Het | 1 | −/− | +/− |
| — | 3-151 | — | Het | 2 | −/− | −/− |
Current report.
Bishop DF, et al, unpublished data.
Increased HFE C282Y gene frequency in hemizygous XLSA probands.
In the analysis of the frequency of HFE mutant alleles in XLSA patients, we have only counted the proband in each family; the person who first presented and was detected with XLSA for whatever reason. In addition to the eight probands analyzed here for the HFE gene mutations, we also analyzed 14 additional XLSA probands from unrelated families for these HFE mutations (data not shown) and found one male proband who was a compound heterozygote for C282Y and H63D, one male heterozygous for C282Y, and four probands heterozygous for H63D: one female and three males (Table 3). To date, we are aware of one additional published study of coinheritance of HFE andALAS2 mutations encompassing a single proband (also summarized in Table 3): a compound heterozygote for C282Y and H63D.36The gene frequencies of the two HFE mutations in all 18 unrelated hemizygous XLSA probands available to date (excluding the Chinese proband of our family 1, since the C282Y mutation is rare or absent in this population,37 and excluding the female heterozygotes, since they are generally at less risk for iron loading) were compared with their frequency in 702 normal individuals matched for country of origin (data compiled from 10 independent studies; Table4). The frequencies were corrected for the fact that there is complete linkage disequilibrium between the C282Y and H63D alleles; if an individual has one of these mutations on a chromosome, the other mutation is never found on that chromosome. Therefore, the number of alleles at risk for C282Y mutations is the total chromosomes minus those carrying H63D and vice versa for the number of alleles at risk for H63D.38 The at-risk allele frequencies of the C282Y and H63D mutations in the chromosomes of unrelated hemizygous XLSA probands were 17.2% and 22.6%, respectively, compared with the at-risk frequencies in the normal white population of 5.5% and 15.3%, respectively. While the total number of XLSA hemizygotes studied was small, there was a threefold increase in the frequency of the C282Y HFE mutation in XLSA chromosomes (Table 4). The chi-square value using the Yates correction for continuity was 4.06 (P = .044), indicating that there was a significant (P ≤ .050) increase in C282Y mutations in hemizygous XLSA chromosomes as compared with those of normal individuals. The H63D mutation was not significantly increased in XLSA hemizygotes (χ2 = 0.097;P = .449).
Comparison of HFE Gene Frequencies in XLSA to Those in Normal Individuals
| Population | XLSA Hemizygotes4-150 | Random Controls4-151 | References | ||||
|---|---|---|---|---|---|---|---|
| Total Chromosomes | C282Y Alleles | H63D Alleles | Total Chromosomes | C282Y Alleles | H63D Alleles | ||
| United Kingdom | 16 | 2 | 4 | 624 | 37 | 81 | 41, 42 |
| United States | 8 | 1 | 2 | 312 | 19 | 49 | 25, 38, 43 |
| Italy | 6 | 1 | 0 | 234 | 2 | 28 | 44, 45 |
| France | 2 | 1 | 1 | 78 | 3 | 13 | 46-48 |
| Germany | 2 | 0 | 0 | 78 | 3 | 11 | 42 |
| The Netherlands | 2 | 0 | 0 | 78 | 2 | 23 | 42 |
| Total: | 36 | 5 | 7 | 1,404 | 66 | 205 | |
| At-Risk allele frequency‡ | 0.172 | 0.226 | 0.055 | 0.153 | |||
| χ2 = | 4.06 | 0.097 | — | — | |||
| P = | .044 | .449 | — | — | |||
| Population | XLSA Hemizygotes4-150 | Random Controls4-151 | References | ||||
|---|---|---|---|---|---|---|---|
| Total Chromosomes | C282Y Alleles | H63D Alleles | Total Chromosomes | C282Y Alleles | H63D Alleles | ||
| United Kingdom | 16 | 2 | 4 | 624 | 37 | 81 | 41, 42 |
| United States | 8 | 1 | 2 | 312 | 19 | 49 | 25, 38, 43 |
| Italy | 6 | 1 | 0 | 234 | 2 | 28 | 44, 45 |
| France | 2 | 1 | 1 | 78 | 3 | 13 | 46-48 |
| Germany | 2 | 0 | 0 | 78 | 3 | 11 | 42 |
| The Netherlands | 2 | 0 | 0 | 78 | 2 | 23 | 42 |
| Total: | 36 | 5 | 7 | 1,404 | 66 | 205 | |
| At-Risk allele frequency‡ | 0.172 | 0.226 | 0.055 | 0.153 | |||
| χ2 = | 4.06 | 0.097 | — | — | |||
| P = | .044 | .449 | — | — | |||
Calculated from patient data listed in Table 3 omitting family 1 and the heterozygotes.
The proportion of normal chromosomes analyzed for the 6 representative populations was kept identical to those for the XLSA populations. The absolute number of chromosomes in each population was set proportional to that for The Netherlands population, which was, relatively, the smallest sample size. The number of C282Y and H63D alleles in each population was calculated using the respective allele frequency found in the particular study.
Calculated as: frequency = [C282Y Alleles]/[(Total Chromosomes) − (H63D Alleles)] and [H63D Alleles]/[(Total Chromosomes) − (C282Y Alleles)], since there is complete linkage disequilibrium between C282Y and H63D.
DISCUSSION
Molecular heterogeneity of ALAS2 mutations in XLSA.
We report four new mutations of the ALAS2 gene from unrelated families with pyridoxine-responsive XLSA. Three were in exon 9 and one was in exon 5. With this report, publications have described 15 unrelated families with XLSA caused by 15 different ALAS2mutations (Table 5). All were missense mutations. Many (29%) of the mutations in unrelated XLSA patients are in CpG dinucleotides, hotspots for mutation apparently resulting from spontaneous deamination of 5-methylcytosine to thymine. While mutations have been found in each exon of the catalytic domain (exons 5-11),39 thus far the preponderance of XLSA probands have mutations in exon 9, the exon containing the PLP binding site (K391). The clustering of mutations in exon 9 may simply be due to the fact that exon 9 has 8 CpG dinucleotides—about twice that of any of the other catalytic domain exons. Alternatively, it suggests that mutations affecting PLP binding are tolerated better than others. Exon 5 has the second greatest number of mutations and is possibly involved in PLP binding since the exon 5 Tyr 199 mutated in family 5 is homologous to the Tyr 70 residue that contacts PLP in aspartate aminotransferase.40 The four mutations reported here all involve highly conserved residues, with both tyrosine 199 and arginine 411 invariant among all reported ALAS isozymes. None of the four mutations were polymorphisms, as evidenced by their lack of occurrence in 100 normal ALAS2 alleles. The mutations segregated with XLSA clinical phenotype within three of the families and between the fourth proband and an unrelated family.
Published Mutations of the ALAS2 Gene in Patients With XLSA
| Exon | Base | Sequence Context | Amino Acid | Mutation Type | PLP5-150 | Age of Onset (yr) | Sex | Reference |
|---|---|---|---|---|---|---|---|---|
| 5 | C547A | TTC → TTA | F165L | Transversion | + | 0 | M | 4 |
| 5 | G561T | CGC → CTC | R170L | Transversion | ++ | 30 | M | 49 |
| 5 | G566A | GCT → ACT | A172T | Transition | ++++ | 81 | F | 6 |
| 5 | A621T | GAT → GTT | D190V | Transversion | − | 18 | M | 8 |
| 5 | T647C | TAC → CAC | Y199H | Transition | ++ | 16 | M | This report |
| 7 | G923A | GGT → GAT | G291S | Transition | +++ | 35 | M | 7 |
| 7 | A947C | AAG → GAG | K299Q | Transition | ++++ | 77 | M | 6 |
| 8 | C1215G | ACT → AGT | T388S | Transversion | ++ | 55 | M | 5 |
| 9 | C1283T | CGC → TGC | R411C | CpG transition | +++ | 8 | M | This report |
| 9 | A1328G | ATG → GTG | M426V | Transition | +++ | 2 | M | 8 |
| 9 | G1395A | CGA → CAA | R448Q | CpG transition | ++ | 11 | M | This report |
| 9 | C1406T | CGC → TGC | R452C | CpG transition | δ | ? | M | This report |
| 9 | G1407A | CGC → CAC | R452H | CpG transition | + | 24 | M | 9 |
| 9 | T1479A | ATC → AAC | I476N | Transversion | +++ | 16 | M | 3 |
| 10 | C1622G | CAC → GAC | H524D | Transversion | ++ | 0 | M | 10 |
| Exon | Base | Sequence Context | Amino Acid | Mutation Type | PLP5-150 | Age of Onset (yr) | Sex | Reference |
|---|---|---|---|---|---|---|---|---|
| 5 | C547A | TTC → TTA | F165L | Transversion | + | 0 | M | 4 |
| 5 | G561T | CGC → CTC | R170L | Transversion | ++ | 30 | M | 49 |
| 5 | G566A | GCT → ACT | A172T | Transition | ++++ | 81 | F | 6 |
| 5 | A621T | GAT → GTT | D190V | Transversion | − | 18 | M | 8 |
| 5 | T647C | TAC → CAC | Y199H | Transition | ++ | 16 | M | This report |
| 7 | G923A | GGT → GAT | G291S | Transition | +++ | 35 | M | 7 |
| 7 | A947C | AAG → GAG | K299Q | Transition | ++++ | 77 | M | 6 |
| 8 | C1215G | ACT → AGT | T388S | Transversion | ++ | 55 | M | 5 |
| 9 | C1283T | CGC → TGC | R411C | CpG transition | +++ | 8 | M | This report |
| 9 | A1328G | ATG → GTG | M426V | Transition | +++ | 2 | M | 8 |
| 9 | G1395A | CGA → CAA | R448Q | CpG transition | ++ | 11 | M | This report |
| 9 | C1406T | CGC → TGC | R452C | CpG transition | δ | ? | M | This report |
| 9 | G1407A | CGC → CAC | R452H | CpG transition | + | 24 | M | 9 |
| 9 | T1479A | ATC → AAC | I476N | Transversion | +++ | 16 | M | 3 |
| 10 | C1622G | CAC → GAC | H524D | Transversion | ++ | 0 | M | 10 |
Abbreviations: M, male; F, female.
Response to pyridoxine supplementation: ++++, complete normalization of Hb and MCV; +++, >4 g/dL increase in Hb; ++, 2-4 g/dL; +, 1-2 g/dL; δ, partial response; −, no response.
The importance of gene-based diagnosis for all relatives of XLSA probands was highlighted by the variation in erythrocyte size dimorphism in heterozygotes due to variable lyonization in family 6 heterozygotes (Fig 2). The carrier female 1.1 was not detected by blood film examination or by measurements of Hb, MCH, MCV, RDW, RBC size histogram, or RBC scattergram (Fig 2D). Without careful evaluation of RBC dimorphism by histogram (Fig 2B and C), scattergram, or blood film, the carrier females II.3 and III.7 would also have been considered normal based on their MCV and MCH values (Table 1).
The Y199H mutation was de novo in XLSA family 5.
The highly informative and intragenic CA dinucleotide repeat30 in intron 7 of the ALAS2 gene made it possible to determine the origin of the T647C (Y199H) mutation in family 5. The T647C mutation in the proband and his mother was on the same chromosome as the A5 allele of the intron 7 CA-repeat, which the proband’s mother was obligated to receive from her father, based on her sisters’ haplotypes (Fig 3B). Since the mutation was present in the proband’s mother’s lymphocytes, it was germline and came from her father’s gamete, which was an isolated meiotic mutation—none of his other five daughters having inherited the ALAS2 mutation. Although this base change was not in a CpG dinucleotide, the presence of numerous CpG hotspots for XLSA mutations may result in additional de novo cases of XLSA and thus ALAS2 mutations should be considered in microcytic sideroblastic anemia even if X-linked inheritance is not complete.
Iron overload compromises pyridoxine responsiveness.
We have previously noted a correlation between dramatically high iron levels and reduced pyridoxine responsiveness in another unrelated proband with XLSA.4 In the present study, long-term monitoring of the hematologic status of family 5 and 6 probands with and without pyridoxine supplementation has implicated iron overload as a complicating factor in assessing pyridoxine responsiveness. The proband in family 6 (mutation R411C) was monitored over a 28-year period (Fig 1B). At presentation (age 8), his transferrin saturation (20%) indicated lack of iron overload and he was quite responsive to low-dose (3 mg/d) pyridoxine with an increase in Hb concentration of approximately 4 g/dL, which was maintained by this low dosage for the following 11 years, during which time transferrin saturation increased to nearly 100%, exacerbated by administration of oral iron. At the end of this time, after iron administration was terminated, he still showed a 4-g/dL oscillation in Hb when temporarily removed from pyridoxine supplementation. However, over the subsequent 10 years, the proband’s Hb concentration steadily decreased during management on 200 to 300 mg/d pyridoxine. This decrease occurred over a time when the patient was becoming more loaded with iron as evidenced by an increase in serum ferritin from 1,000 to greater than 3,000 μg/L and was starting to show clear evidence of tissue damage as indicated by glucose intolerance and incipient cardiac disease. That the patient was able to maintain a much higher Hb level when his iron stores were lower, even on 3 mg/d pyridoxine, is suggestive of a damaging effect of iron loading on erythropoiesis and/or heme biosynthesis.
The proband in family 5 (mutation Y199H) also maintained a higher Hb level when he was taking pyridoxine and his iron stores were at their lowest following chelation therapy (70 months; Fig 1A). In the following 15 months without chelation, storage iron increased and Hb levels decreased, even though pyridoxine supplementation continued. Iron removal by phlebotomy led to an increase in Hb concentration, MCH, and MCV values only when iron stores were reduced to the normal range as indicated by both serum ferritin and transferrin saturation levels (Fig 1A). High iron may compromise mitochondrial function and hence heme biosynthesis. Yeast with a mutation in the Atm1p transporter accumulate 30-fold more iron in their mitochondria than wild-type cells and show elevated glutathione and H2O2hypersensitivity, indicating oxidative stress.50 It also may be relevant that high ferrous iron concentrations inhibit ALAS activity in vitro.51 These results suggest that pyridoxine responsiveness and/or Hb synthesis can be blocked by iron overload and highlight the importance of appropriate patient management to prevent or reverse iron overload. Patients who present with iron overload should not be considered refractory to pyridoxine therapy until iron stores are normalized with serum ferritin and transferrin saturation in the normal range. Our studies demonstrate that phlebotomy alone is sufficient to reverse iron overload of moderate degree. Of further interest is the success of daily pyridoxine and quarterly phlebotomy in maintaining the proband of family 8 (R452C) with no further incidence of anemia during the last 18 years. It should be recognized that although a patient may have moderate anemia, it is not counterproductive to phlebotomize, as our experience demonstrates that Hb typically increases following blood removal, rather than decreases (Fig 1A). This tolerance of chronic phlebotomy in sideroblastic anemia patients, frequently with elevation in Hb once iron stores are depleted, has been well documented.52-56
Coinheritance of HH may exacerbate XLSA.
The iron-loading disorder, HH, has recently been discovered to be caused by mutations (C282Y and H63D) in an HLA-H gene now designatedHFE.25,57 Both mutations are functionally similar in increasing transferrin binding by the transferrin receptor resulting in increased iron uptake.57 Iron accumulation and storage in both XLSA and HH share a similar picture of clinical pathology, with both involving slowly progressing accumulation of iron in various tissues, both resulting in increased transferrin saturation and increased serum ferritin, and both leading to the same clinical pathologies of rheumatoid arthritis, growth disturbances, diabetes, liver cirrhosis, and heart failure due to toxic iron concentrations. Coinheritance of XLSA and HH mutations might therefore be predicted to result in more rapid increases in iron stores and thus earlier development and detection of pathology in individuals who might otherwise be unrecognized if their XLSA was mild. Analysis (Table 4) of 18 unrelated hemizygous XLSA probands diagnosed in our laboratories, including one additional published case, showed a significant (P = .044, χ2 test) threefold increase in allele frequency of the C282Y mutation in this patient group’s chromosomes compared with those of normal individuals when matched for country of origin. However, it must be stressed that the total number of patients analyzed was small, and the power of this statistic to test the correlation was low in both cases. Nonetheless, the results clearly support the suggestion that coinheritance of the HFEC282Y allele is likely increased in those XLSA patients coming to clinic and should also be considered an additional risk factor for development of pathology in XLSA patients.
Uniquely, the proband in family 5 was both hemizygous for theALAS2 mutation Y199H and homozygous for the HFEmutation C282Y, and was heavily iron loaded already at presentation at age 16 with 100% transferrin saturation and serum ferritin greater than 1,000 μg/L. The proband’s mother did not show any signs of increased iron, but should be monitored after menopause due to the increased risk of iron loading, since she is heterozygous for both XLSA and C282Y (Fig 3C). It is likely that differences in severity of XLSA genotypes, varying degrees of lyonization of the mutant X chromosome, and differences in blood loss due to menstruation and child birth will complicate assessment of contributions of iron overload fromHFE alleles in XLSA heterozygotes. It may be of importance that among seven XLSA heterozygotes, including one also heterozygous for theHFE C282Y mutation, only XLSA heterozygote II.3 in family 6, who was homozygous for the H63D mutation, showed increased iron stores. Her mean transferrin saturation was 55% (range, 39% to 82%), compared with her mother and daughter (13% and 38%, respectively). Her serum ferritin was also above normal on 8 of 12 occasions (Table1).
Additional evidence for increased iron stores in pyridoxine-responsive XLSA with coinheritance of HH was provided by a family in which two brothers presented with XLSA, aged 59 and 66 years.36 The younger brother had twice the body iron stores as the older and was a compound HH heterozygote, while the older brother had neitherHFE mutation. Recent studies based on direct analysis of the C282Y and H63D mutations have provided additional support for the iron-loading propensity of these mutations in the heterozygous state. Roberts et al58 and Santos et al59 found an increased frequency of the C282Y allele in patients with porphyria cutanea tarda, another iron-loading disorder. Other studies show that the mean serum ferritin concentration and percent transferrin saturation was higher in HFE heterozygotes60-62 and in HFE compound heterozygotes48 than in normal individuals. As detection of XLSA patients improves and the database of clinical data increases, it may be possible to estimate the increased risk of iron overload in XLSA patients with coinheritance ofHFE mutations, but it will always be best to individually monitor and treat XLSA family members for iron elevation to minimize the risk of diabetes, arthritis, and/or other disorders as these individuals age.
Pyridoxine responsiveness in XLSA.
All four mutations described here resulted in pyridoxine-responsive anemia, with the variable response of Hb, MCV and MCH to pyridoxine therapy in families 5 through 8 apparently due not only to genetic heterogeneity, but also to environmental factors. Pyridoxine responsiveness may appear to decrease in old age due to decreased pyridoxine bioavailability.63 64 As shown in this study, differences in iron loading also effect pyridoxine responsiveness. This can be seen in the reduced pyridoxine responsiveness with iron loading for the probands of families 5 and 6 (Fig 1), as well as in family 7, where Hb steadily decreased from 11.9 to 10.3 g/dL during 2 years on pyridoxine supplementation. As noted earlier, coinheritance ofHFE mutation alleles likely also contributes to variations in iron loading and thus in pyridoxine responsiveness.
Notably, all previously published XLSA families in which the mutation has been determined have been pyridoxine-responsive (Table 5), save the D190V mutation, which results in an enzyme that is altered in its posttranslational processing and which is only 5% of normal enzyme activity in bone marrow. In this case, one would not expect much of a clinical effect, even if the small amount of residual enzyme was activated by pyridoxine. Pyridoxine-refractory hereditary sideroblastic anemia could also be due to causes other than ALAS2 gene mutations and we previously reported exclusion of X-linkage for one such case with evidence for autosomal inheritance.65
The sometimes small responses to pyridoxine therapy raise the question of what constitutes a pyridoxine response. We propose that any statistically significant increase in Hb level while taking pyridoxine should be sufficient to categorize a patient as responsive. To ensure that this response is truly related to the pyridoxine treatment requires repeated observation of this response by withdrawing the pyridoxine for a certain period of time and then restarting. As stated earlier, one should not give up on such analyses until the iron overload has been eliminated—preferably by phlebotomy and occasionally with concomitant desferrioxamine if iron stores are threatening organ failure. A complicated clinical picture should not deter researchers from attempting this repetition, since a measurable response is likely to be followed by a recommendation to the patient of lifelong supplementation with doses of pyridoxine in excess of what is normally available in the diet along with occasional phlebotomy to manage the undiminished propensity to iron loading due to ineffective erythropoiesis. Early diagnosis and management to maintain normal iron levels are now possible for individuals with XLSA and should be pursued for all family members as it is possible that, as with HH, these efforts can result in an essentially normal lifespan.66
ACKNOWLEDGMENT
Permission to review the clinical data of their patients was kindly provided by Dr J.K. Whittaker, Prof A.K. Burnett, University Hospital of Wales, Wales, UK, Dr Paula Cotter, University Hospital of Cork, Eire, and Dr D. Samson, Charing Cross and Westminster Medical School, University of London, London, UK. We thank Dr C.E. McLaren for the analysis of RBC size distributions (Fig 2) and other clinicians who referred patient sample for diagnosis. The authors thank A. Leahy, M. Piccione, and V. Tchaikovskii for expert technical assistance.
Supported in part by Grant No. (R01 DK40895) from the National Institutes of Health and No. (584) from the March of Dimes Birth Defects Foundation to D.F.B.; Scottish Home and Health Department Grant No. K/MRS/50/C2012 to E.J.F.; and grants from the IRCCS Policlinico S. Matteo, University of Pavia, and MURST to M.C. P.D.C. was the recipient of a March of Dimes Birth Defects Foundation Predoctoral Graduate Research Training Fellowship.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. section 1734 solely to indicate this fact.
REFERENCES
NOTE ADDED IN PROOF
The family 6 mutation, R411C, was reported in an unrelated family while this article was in press and showed similar hemoglobin levels and responsiveness to pyridoxine.67

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal