

Key Points
Emergency granulopoiesis during peritonitis evoked changes in BM resolvins.
RvD4 regulates emergency granulopoiesis and excessive neutrophil infiltration at the site of infection at the single-cell level.

Abstract
Neutrophils reside in the bone marrow (BM), ready for deployment to sites of injury/infection, initiating inflammation and its resolution. Here, we report that distal infections signal to the BM via resolvins to regulate granulopoiesis and BM neutrophil deployment. Emergency granulopoiesis during peritonitis evoked changes in BM resolvin D1 (RvD1) and BM RvD4. We found that leukotriene B4 stimulates neutrophil deployment. RvD1 and RvD4 each limited neutrophilic infiltration to infections, and differently regulated BM myeloid populations: RvD1 increased reparative monocytes, and RvD4 regulated granulocytes. RvD4 disengaged emergency granulopoiesis, prevented excess BM neutrophil deployment, and acted on granulocyte progenitors. RvD4 also stimulated exudate neutrophil, monocyte, and macrophage phagocytosis, and enhanced bacterial clearance. This mediator accelerated both neutrophil apoptosis and clearance by macrophages, thus expediting the resolution phase of inflammation. RvD4 stimulated phosphorylation of ERK1/2 and STAT3 in human BM-aspirate–derived granulocytes. RvD4 in the 1 to 100 nM range stimulated whole-blood neutrophil phagocytosis of Escherichia coli. RvD4 increased BM macrophage efferocytosis of neutrophils. Together, these results demonstrate the novel functions of resolvins in granulopoiesis and neutrophil deployment, contributing to the resolution of infectious inflammation.
Introduction
Human neutrophil deployment from the bone marrow (BM) to the peripheral blood is critical for homeostasis, the initiation of the acute inflammatory response, and its self-limited resolution.1-3 BM is a lipid-rich compartment in which hematopoietic stem and progenitor cells (HSPCs) reside,2 and is the primary site of granulopoiesis. HSPCs are tightly regulated to continuously replenish billions of myeloid cells daily that are necessary to maintain physiological cell numbers in circulation.2,4 The acute inflammatory response is the body’s first line of defense against invading pathogens. Acute inflammatory signals associated with infection lead to increase(s) in BM granulopoiesis and neutrophil deployment to contain invaders.2 Failure to control local infection can lead to systemic bacterial dissemination, as in sepsis, evoking the BM to switch to an emergency state.1,4
Resolution of self-limited acute inflammatory response is an active process regulated in part by production of endogenous specialized proresolving mediators (SPMs) that orchestrate the return to homeostasis.3 SPMs comprise structurally distinct families of lipid mediators including arachidonic acid–derived lipoxins (LXs), eicosapentaenoic acid (EPA)–derived E-series resolvins (Rvs), docosahexaenoic acid (DHA) –derived D-series Rv, protectins (PD), and maresins. Each SPM exhibits cell-type–specific actions in immune responses,3,5 activates the resolution of acute inflammation, and they are resolution agonists that function as immunoresolvents. SPMs play a pivotal role in regulating host defense mechanisms in neutrophils via potently enhancing microbial containment and phagocytosis, regulating trafficking into the inflammatory-infectious site, and stimulating clearance of apoptotic leukocytes and cellular debris by macrophages.3,5
Structures of several of the SPMs proved to be conserved in evolution. For example, LXA4 biosynthesis from arachidonic acid and LXA5 from precursor EPA were identified in the head kidney gland of trout, which is the hematopoietic organ equivalent to mammalian BM.6,7 Both PD1 and resolvin D1 (RvD1) are biosynthesized by trout,8 and LXA4 is present during infections in zebrafish.9 Human BM produces LXA4 and LXB4 that stimulates BM colony-forming units (CFU)10 at concentrations as low as 10−10 M. RvD4 was recently identified in both human and mouse BM.11 Hence, the production and potential function(s) of SPMs in BM are of interest. Along these lines, neuroprotectin D1 (NPD1/PD1), also biosynthesized from DHA,12 promotes cardiac and neuronal differentiation,13 in which injury and inflammation can impact both differentiation and proliferation of stem cells via regulation of the local oxidative environment.13 Given that myeloid stem and progenitor cells reside in the BM, we hypothesized that there may be a relationship between the resolution phase of infectious exudates and the host’s deployment of polymorphonuclear neutrophils (PMN). Here, we report that self-limited infections evoke time-dependent changes in BM lipid mediators that regulate excessive granulopoiesis and neutrophil deployment.
Methods
Human BM and whole-blood neutrophils
Fresh BM aspirates from deidentified individuals were purchased from AllCells, Inc (Alameda, CA) with Mass General Brigham investigational review board protocol number 1999P0001279. For information on donor demographics, see supplemental Table 1, available on the Blood website. Human peripheral blood was drawn from healthy volunteers (Mass General Brigham investigational review board protocol number 1999-P-001297) by venipuncture with sodium heparin. Whole blood was aliquoted for neutrophil phagocytosis (see supplemental Methods).
Self-limited Escherichia coli peritonitis
Animal experimental procedures were approved by the institutional animal care and use committee of Brigham and Women’s Hospital (protocol number 2016N000145). Male Friend leukemia virus B (FVB) mice (6- to 8-weeks-old, from Charles River, Wilmington, MA) were inoculated intraperitoneally with live E coli serotype O6:K2:H1 at 105 CFUs per mouse, in sterile saline, as reviewed in Serhan.3 RvD4 (100 ng per mouse), RvD1 (100 ng per mouse), or vehicle (0.01% ethanol) was administered IV to mice. For a separate set of experiments, we administered RvD4 (IV) at concentrations of 1, 10, or 100 ng per mouse, or vehicle only (0.01% ethanol), to mice with peritonitis.
At the designated time intervals, mice were euthanized, and exudates, whole blood, and BM cells were collected. Information on SPM-targeted metabololipidomics profiling, single-cell mass cytometry, flow cytometry, enzyme-linked immunosorbent assay, and bacterial titers is presented in the supplemental Methods.
Authentication of RvD1 and RvD4
RvD1 (CAS # 872993-05-0) and RvD4 (CAS # 1025684-60-9) were purchased from Cayman Chemical; each was authenticated to match the physical properties with our original material (RvD114 and RvD411). The integrity of RvD1 and RvD4 was assessed using UV spectroscopy and liquid chromatography–tandem mass spectrometry (LC-MS/MS) to obtain retention times, and MS/MS spectra for direct comparisons.11,14
Statistics
Statistical analyses were performed using a 2-tailed Student t test for 2 group comparisons, 1-way analysis of variance (ANOVA) with multiple group comparisons for ≥3 independent groups, or 2-way ANOVA with multiple group comparisons for 2 biological variables (GraphPad Prism). P < .05 was statistically significant.
Results
BM: Rvs and changes in myeloid cells during remote infections
Because BM is the site of granulopoiesis,2 we questioned whether self-resolving bacterial infections affect BM proresolving mediators and neutrophil deployment. Results in Figure 1 demonstrate that bacterial peritonitis evokes temporal changes in BM-SPMs and emergency granulopoiesis. Intraperitoneal inoculation of E coli (105 CFU)3 initiated self-limited acute infections. Peritonitis-initiated neutrophil (PMN) influx, identified as CD45+CD11b+Ly6G+ F4/80−Ly6C− by flow cytometry (Figure 1A; supplemental Figure 1A), reached a maximum (Ψmax) at 12 hours (Tmax), followed by a decline to 50% (Ψ50) at 30 hours (T50). This gave a resolution interval (Ri) of 18 hours (T50 − Tmax), marking the resolution phase of inflammation (Figure 1A). Peritonitis also triggered a time-dependent increase in circulation and BM neutrophils (Figure 1B-C; supplemental Figure 1B), along with granulocyte progenitors (Figure 1D; supplemental Figure 1C-D). This natural resolution of infectious inflammation is an example of temporal changes in cellular composition in exudates during the time course of infection from initiation to resolution of the acute inflammatory response.
![Temporal changes in neutrophils during infection. Mice were inoculated intraperitoneally (IP) with E coli (105 CFU, self-limited dose; see “Methods”). Exudates and BM cells were collected at 0, 12, 24, 48, and 72 hours after inoculation. Numbers of exudate (A); whole blood (B); BM neutrophils (C); and BM granulocyte progenitors, LSKs and GMPs (D), were determined by flow cytometry. (E) Screen captures of RvD1 from targeted LC-MS/MS analysis. (Top) BM RvD1–targeted scheduled multiple reaction monitoring (MRM) in negative-ion mode for mass-to-charge (m/z) 375 → 215 (“→” indicates fragmentation/transition). (Bottom) Synthetic RvD1. MRM signal-to-noise ratio of >80. (F) Screen captures of RvD4 from targeted LC-MS/MS analysis. (Top) BM RvD4–targeted scheduled MRM in negative-ion mode for m/z 375 → 101. (Bottom) Synthetic RvD4. MRM signal-to-noise ratio of >40. (G) Screen capture of BM RvD4 MS/MS fragmentation. Inset: RvD4 structure and proposed fragmentation. BM RvD4 MS/MS fragmentation spectrum matched to an unbiased library of synthetic RvD4 standard with a fit score of 94.5%. BM-RvD4 MS/MS spectral ions gave a parent ion mass of m/z 375 (M-H) and daughter ions of m/z 357 (M-H–H2O), 339 (M-H–2H2O), 313 (M-H–H2O-CO2), 295 (M-H–2H2O-CO2), 255 (273–H2O), 233 (277–CO2), and 101 (M-H–CHOH-[CH]6-CH2-[CH]4-CHOH-CH2-[CH]2-CH2-CH3). (E-F) Screen captures taken from Sciex OS version 1.7.036606 (Explorer Mode). Note the additional digits in the screen captures in panel G are the default setting (the number [amu] in the second decimal place does not accurately reflect the mass of the ions).](/view-large/figure/11799700/BLOOD_BLD-2022-019145-gr1.jpg)
Temporal changes in neutrophils during infection. Mice were inoculated intraperitoneally (IP) with E coli (105 CFU, self-limited dose; see “Methods”). Exudates and BM cells were collected at 0, 12, 24, 48, and 72 hours after inoculation. Numbers of exudate (A); whole blood (B); BM neutrophils (C); and BM granulocyte progenitors, LSKs and GMPs (D), were determined by flow cytometry. (E) Screen captures of RvD1 from targeted LC-MS/MS analysis. (Top) BM RvD1–targeted scheduled multiple reaction monitoring (MRM) in negative-ion mode for mass-to-charge (m/z) 375 → 215 (“→” indicates fragmentation/transition). (Bottom) Synthetic RvD1. MRM signal-to-noise ratio of >80. (F) Screen captures of RvD4 from targeted LC-MS/MS analysis. (Top) BM RvD4–targeted scheduled MRM in negative-ion mode for m/z 375 → 101. (Bottom) Synthetic RvD4. MRM signal-to-noise ratio of >40. (G) Screen capture of BM RvD4 MS/MS fragmentation. Inset: RvD4 structure and proposed fragmentation. BM RvD4 MS/MS fragmentation spectrum matched to an unbiased library of synthetic RvD4 standard with a fit score of 94.5%. BM-RvD4 MS/MS spectral ions gave a parent ion mass of m/z 375 (M-H) and daughter ions of m/z 357 (M-H–H2O), 339 (M-H–2H2O), 313 (M-H–H2O-CO2), 295 (M-H–2H2O-CO2), 255 (273–H2O), 233 (277–CO2), and 101 (M-H–CHOH-[CH]6-CH2-[CH]4-CHOH-CH2-[CH]2-CH2-CH3). (E-F) Screen captures taken from Sciex OS version 1.7.036606 (Explorer Mode). Note the additional digits in the screen captures in panel G are the default setting (the number [amu] in the second decimal place does not accurately reflect the mass of the ions).
Temporal changes in neutrophils during infection. Mice were inoculated intraperitoneally (IP) with E coli (105 CFU, self-limited dose; see “Methods”). Exudates and BM cells were collected at 0, 12, 24, 48, and 72 hours after inoculation. Numbers of exudate (A); whole blood (B); BM neutrophils (C); and BM granulocyte progenitors, LSKs and GMPs (D), were determined by flow cytometry. (E) Screen captures of RvD1 from targeted LC-MS/MS analysis. (Top) BM RvD1–targeted scheduled multiple reaction monitoring (MRM) in negative-ion mode for mass-to-charge (m/z) 375 → 215 (“→” indicates fragmentation/transition). (Bottom) Synthetic RvD1. MRM signal-to-noise ratio of >80. (F) Screen captures of RvD4 from targeted LC-MS/MS analysis. (Top) BM RvD4–targeted scheduled MRM in negative-ion mode for m/z 375 → 101. (Bottom) Synthetic RvD4. MRM signal-to-noise ratio of >40. (G) Screen capture of BM RvD4 MS/MS fragmentation. Inset: RvD4 structure and proposed fragmentation. BM RvD4 MS/MS fragmentation spectrum matched to an unbiased library of synthetic RvD4 standard with a fit score of 94.5%. BM-RvD4 MS/MS spectral ions gave a parent ion mass of m/z 375 (M-H) and daughter ions of m/z 357 (M-H–H2O), 339 (M-H–2H2O), 313 (M-H–H2O-CO2), 295 (M-H–2H2O-CO2), 255 (273–H2O), 233 (277–CO2), and 101 (M-H–CHOH-[CH]6-CH2-[CH]4-CHOH-CH2-[CH]2-CH2-CH3). (E-F) Screen captures taken from Sciex OS version 1.7.036606 (Explorer Mode). Note the additional digits in the screen captures in panel G are the default setting (the number [amu] in the second decimal place does not accurately reflect the mass of the ions).
To determine the impact of peritoneal infections on BM-SPMs, we collected BM samples (Figure 1A) and subjected them to targeted metabololipidomics. BM-SPMs were identified and quantified, using rigorous matching criteria based on each SPM's unique physical and chemical properties (see supplemental Methods). RvD1 in the BM was identified by matching its retention time of 11.5 minutes (Figure 1E, upper panel) to that of synthetic RvD1 (Figure 1E, lower panel). The MS/MS fragmentation spectra of BM RvD1 (supplemental Figure 1E) were consistent with the published physical properties reported for RvD1.14
In bacterial infections, RvD3 and RvD4 appear late in the resolution phase of infectious inflammation exudates.15,16 Endogenous RvD4 was also identified in the BM, eluting at 12.4 minutes along with 2 other products at 12.7 minutes (denoted I) and 12.9 minutes (denoted II) (Figure 1F, upper panel). The signal at the retention time of 12.4 minutes demonstrated an identical chromatography behavior as that of RvD4 (Figures 1F, lower panel). The other 2 products, I and II, proved to be nonenzymatic trans-triene isomers of RvD4 (denoted 10E-RvD4 and10E,13E-RvD4, respectively).11These 2 isomers are produced from the 4S,5S-epoxy-Rv intermediate (4S,5S-epoxy-17S-hydroxy- 6E,8E,10Z,13Z,15E,19Z-docosahexaenoic acid) via nonenzymatic hydrolysis, whereas RvD4 is the proposed enzymatic product.11,17 These 2 isomers, 10E-RvD4 and 10E,13E-RvD4, are essentially not bioactive.11 The BM-RvD4 MS/MS spectra (Figure 1G) essentially matched those reported.11 RvD5, 17-HDHA, RvE1, and 18-HEPE were also identified in the BM (supplemental Figure 1G-J).
Next, we determined whether BM-SPM levels were modulated during peritonitis. RvD1 levels (63.6 ± 14.7 pg per 106 BM cells) were ∼10 times higher at 12 hours compared with levels at 0 hours (6.3 ± 1.3 pg per 106 BM cells) and remained elevated at 24 to 48 hours (Figure 2A). It is noteworthy that RvD4 (13.1 ± 0.6 pg per 106 BM cells) had a statistically significant decrease (P < .001) at 12 hours (supplemental Figure 1F) from basal levels at 0 hours, and the nadir was at 72 hours (Figure 2A). Between 24 to 48 hours, a statistically significant decrease in RvD5 was observed compared with amounts at 12 hours. Importantly, RvD1, RvD4, RvD5, 17-HDHA, RvE1, and 18-HEPE were all identified in the BM at time 0. RvD1, RvD4, and RvD5 showed temporal changes in the BM during peritoneal infections, whereas no apparent significant change was observed in EPA-derived mediators (eg, 18-HEPE and RvE1; Figure 2A). We submitted the identified BM-SPMs to unsupervised principal component analysis. The 3D score plot (Figure 2B, left panel) and the loading plot (Figure 2B, right panel) denote distinct time-dependent clustering of BM-Rvs during distal infections. Specifically, RvD4 strongly associated with time 0 and distinctly separated from other DHA- and EPA-derived Rvs and pathway markers. In circulation, a statistically significant decrease in RvD4 (P < .001) (supplemental Figure 1K-L) (Figure 2C) was observed during the time course of peritonitis.

Time-dependent changes in BM Rvs. (A) Time course of D-series Rvs and proresolving mediators in the BM from targeted LC-MS/MS. (B) Principal component analysis of time course of BM Rvs and proresolving mediators; (left) 3D scoring plot, and (right) 3D loading plot. Results are expressed as mean ± standard error of the mean (SEM); n = 4 or 6 mice per time point, panels A-B. Panel A: time 0 vs 12, 24, 48, and 72 hours, respectively; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. Panel A: time 0 vs 12, 24, 48, and 72 hours, respectively, ∗P < .05, ∗∗∗∗P < .0001; 12 hours vs 48 and 72 hours, respectively, #P < .05, ####P < .0001; 48 vs 72 hours, ‡P < .05. (C) Time course of RvD4 in whole blood as determined with targeted LC-MS/MS. RvD4 blood pooled from 3 mice for each time point. Time 0 vs 12, 24, 48, and 72 hours, respectively; ∗∗∗∗P < .0001. Statistical analysis was carried out using 1-way ANOVA with Bonferroni multiple comparison test. (D-F) CyTOF: BM (CD45+CD41−) isolated from mice with peritonitis at 0, 12, and 72 hours. (D) Composite BM map: force-directed layout (Vortex) showing (94 500 total single cells) clustered by X-shift (n = 3 mice per time point: 0, 12, and 72 hours; ∼10 500 cells per mouse). (E) BM time-course maps: force direct layout. (F) Number of BM neutrophil lineage (PN, preneutrophil; IN, immature neutrophil; and MN, mature neutrophils) and granulocyte stem and progenitor cells (MPP3 and GMPs). Results are mean ± SEM, n = 3 mice per time point. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < 0. 0001. Statistical analysis was performed using 1-way ANOVA with Tukey multiple comparison test.
Time-dependent changes in BM Rvs. (A) Time course of D-series Rvs and proresolving mediators in the BM from targeted LC-MS/MS. (B) Principal component analysis of time course of BM Rvs and proresolving mediators; (left) 3D scoring plot, and (right) 3D loading plot. Results are expressed as mean ± standard error of the mean (SEM); n = 4 or 6 mice per time point, panels A-B. Panel A: time 0 vs 12, 24, 48, and 72 hours, respectively; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. Panel A: time 0 vs 12, 24, 48, and 72 hours, respectively, ∗P < .05, ∗∗∗∗P < .0001; 12 hours vs 48 and 72 hours, respectively, #P < .05, ####P < .0001; 48 vs 72 hours, ‡P < .05. (C) Time course of RvD4 in whole blood as determined with targeted LC-MS/MS. RvD4 blood pooled from 3 mice for each time point. Time 0 vs 12, 24, 48, and 72 hours, respectively; ∗∗∗∗P < .0001. Statistical analysis was carried out using 1-way ANOVA with Bonferroni multiple comparison test. (D-F) CyTOF: BM (CD45+CD41−) isolated from mice with peritonitis at 0, 12, and 72 hours. (D) Composite BM map: force-directed layout (Vortex) showing (94 500 total single cells) clustered by X-shift (n = 3 mice per time point: 0, 12, and 72 hours; ∼10 500 cells per mouse). (E) BM time-course maps: force direct layout. (F) Number of BM neutrophil lineage (PN, preneutrophil; IN, immature neutrophil; and MN, mature neutrophils) and granulocyte stem and progenitor cells (MPP3 and GMPs). Results are mean ± SEM, n = 3 mice per time point. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < 0. 0001. Statistical analysis was performed using 1-way ANOVA with Tukey multiple comparison test.
To investigate infection-induced granulopoiesis, we mapped BM cells during self-limited infections using single-cell cytometry by time-of-flight mass spectrometry (CyTOF), which permits multidimensional analysis of immune populations.18 BM cells were obtained from mice with peritoneal infections at 0, 12, and 72 hours (ie, the initiation phase, 0-12 hours) and during the resolution phase (12-72 hours) of acute inflammation. CD45+CD41− BM cells (supplemental Figure 1N) were analyzed using a clustering algorithm X-shift19 that resulted in the annotation of 24 BM immune populations (Figure 2D), identified using surface markers (supplemental Figure 1O; supplemental Table 2). In Figure 2E, time-dependent changes in BM immune populations were denoted. In this inflammation-resolution time course, distal infection evoked an increase in preneutrophils and immature and mature neutrophils in the BM, along with an increase in granulocyte monocyte progenitors (GMPs) and the multipotent myeloid progenitor 3 (MPP3) (Figure 2F). At 72 hours, the percentage of MPP3 remained elevated compared with that at time 0 (Figure 2F). BM MPP3 (MPPG/M, myeloid bias) comprises multipotent progenitors that give rise to the GMPs and contribute to granulopoiesis.20
RvD1 and RvD4 selectively regulate BM myeloid cells
Given that the BM is the main site of granulopoiesis,2 and we observed that peritoneal infections initiate emergency granulopoiesis (Figures 1 and 2), we questioned whether RvD1 and/or RvD4 regulate neutrophil deployment and differentiation in the BM. RvD1 and RvD4 were selected for further interrogation because they showed temporal changes in the BM (Figure 2). Here, RvD1 or RvD4 was separately administered (IV) to mice with self-limited peritoneal infections. BM and exudates were collected at 0, 12, and 72 hours (Figure 3A). The integrity of RvD1 (supplemental Figure 2A) and RvD4 (supplemental Figure 2B) was authenticated by monitoring their physical properties using targeted LC-MS/MS and UV before each experiment, matching the properties to those reported.11,14 In Figure 3B, systemic administration of RvD1 or RvD4 to mice with peritonitis at 12 hours reduced PMN infiltration by 38.1% ± 1.1% and 61.8% ± 3.6%, respectively. Next, BM-CD45+CD41− were analyzed using uniform manifold approximation and projection (UMAP) together with FlowSOM,21 giving the identification of 15 immune cell populations (Figure 3C) based on their surface markers (supplemental Figure 2C; supplemental Table 2).
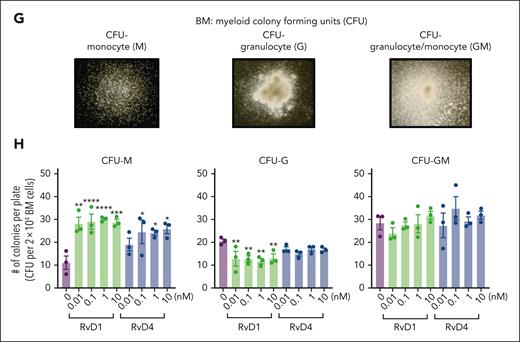
RvD1 and RvD4 regulate BM myeloid cell production during peritonitis. Mice inoculated with E coli (105 CFU, IP, a self-limited dose; see “Methods”) were administered with RvD1 (100 ng per mouse, IV), RvD4 (100 ng per mouse, IV), or vehicle (0.01% v/v ethanol in saline). Exudates and BM cells were collected at 0 hours, 12 hours (initiation phase), and 72 hours (resolution phase) of the acute inflammatory response. (A) Schematic of the experimental design and sample collection. (B) Cell number of neutrophils in the exudate. (C) CyTOF: BM UMAP labeled with 15 immune populations. (D) The number of BM neutrophils, GMPs, LSKs, and Ly6Clow monocyte cells. Results in panels B-D are mean ± SEM, n = 3 mice per time point. Time 0 hour vs 12 and 72 hours; #P < .05, ##P < .01. Rvs (RvD1 or RvD4) vs E coli + vehicle; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001. Statistical analysis was carried out using 2-way ANOVA with Tukey multiple comparison test. (E) BM UMAPs of log fold-change (logFC) between time 0 vs time points (12 or 72 hours), or treatments (RvD1 or RvD4) calculated using edgeR with diffcyt. (F) BM UMAPs of logFC between E coli + RvD4 vs E coli + vehicle calculated using edgeR with diffcyt. (E-F) Only statistically significant populations are colored (P < .05) and adjusted using a Benjamini-Hochberg correction. (G) Light microscopy photographs of BM myeloid CFUs: M, monocytes (left); G, granulocyte (middle), and GM, macrophage/granulocyte (right), obtained with a 2× objective; representative images. (H) BM myeloid CFU quantification after dose-dependent treatment with vehicle only (0.01% ethanol v/v) or with either RvD1 or RvD4 (0.01-10 nM). CFU counts are per 2 × 104 BM cells. Vehicle vs 0.01, 0.1, 1, or 10 nM; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. Statistical analysis was carried out using 2-way ANOVA with Bonferroni multiple comparisons test.
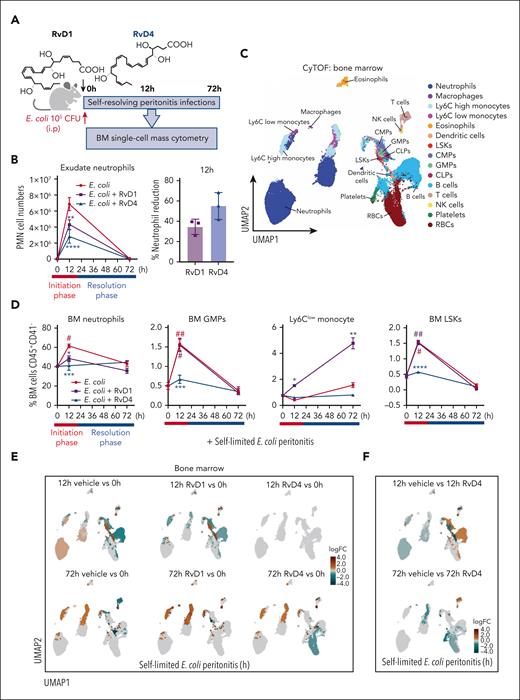
At 12 hours, RvD1-treated mice had decreased BM neutrophils (48.5% ± 5.1%, P < .01) compared with that in mice treated with vehicle only (61.5% ± 3.3%). In comparison, RvD4-treated mice had a statistically significant decrease in BM neutrophils (41.3% ± 2.6%, P < .001), whereas PMN numbers remained nearly the same as at 0 hours (40.3% ± 0.8%) (Figure 3D). RvD4-treated mice had a statistically significant decrease in lineage−Sca1+cKit+ cells (LSKs) (P < .001) and GMPs (P < .0001) at 12 hours compared with vehicle-treated mice, with values essentially the same as those at time 0. In contrast, the numbers of GMPs and LSKs were not altered in RvD1-treated mice compared with vehicle-treated mice (Figure 3D). Because GMPs are also the progenitors for monocyte populations, we determined whether either RvD1 or RvD4 has an impact on these populations. Neither RvD1 nor RvD4 altered the numbers of Ly6Chigh monocytes during peritonitis (supplemental Figure 2D). Of interest, RvD1 increased reparative Ly6Clow monocytes at 12 and 72 hours (Figure 3D) along with an increase in dendritic cells (supplemental Figure 2D) compared with levels in vehicle- or RvD4-treated mice.
Next, by performing differential abundance analysis using the computational framework diffcyt, we determined whether RvD1 or RvD4 plays a role in initiating emergency granulopoiesis.22 BM from RvD4-treated peritonitis mice gave values essentially the same as those obtained at 0 hours, indicating that RvD4 stopped neutrophil deployment and increased LSKs and GMPs in response to distal infections (Figure 3E). RvD4 at 12 hours caused a decrease in LSKs, GMPs, and neutrophils compared with levels in the BM of vehicle-treated mice (Figure 3F). At 72 hours, RvD1 resulted in an increase in Ly6Clow monocytes (supplemental Figure 2E), and RvD4 in an increase in T and B cells (Figure 3F), compared with levels in vehicle-treated mice.
Next, we assessed whether RvD1 or RvD4 regulates myeloid differentiation using a BM CFU assay. Naïve BM cells were incubated with either RvD1 (0.01-10 nM), RvD4 (0.01-10 nM), or vehicle only for 12 days. Colonies were identified and quantified based on their morphological appearance (Figure 3G) as: CFU-monocyte (M), CFU-granulocyte (G), and CFU-granulocyte/macrophages (GM). Both RvD1 and RvD4 resulted in a statistically significantly increase in the numbers of CFU-M in a dose-ependent manner. RvD1 resulted in a statistically significant decrease in the numbers of CFU-G (Figure 3H). In contrast, RvD4 did not elicit statistically significant changes in CFU-G numbers or CFU-GM when compared with those in vehicle- or RvD1-treated colonies (Figure 3H).
RvD4 signals resolution programs in vivo
Because RvD4 is decreased in the BM during self-limited infections and had potent actions in preventing excessive BM neutrophil deployment, we sought evidence for RvD4’s function(s) on neutrophil trafficking during bacterial infections. RvD4 was administered (IV) to mice with self-limited infections, and exudates, whole blood, and BM cells were collected at 0, 12, and 72 hours (Figure 4A). UMAP-FlowSOM21 analysis of CD45+CD41 (supplemental Figure 3A) revealed 17 distinct cell populations in 3 compartments (Figure 4B; supplemental Figure 3B). RvD4-treated mice with peritonitis had reduced numbers of infiltrated neutrophils in the peritoneal cavity (17.9% ± 8.4%, P < .001; left panel exudate) and reduced neutrophil deployment from the BM (Figure 4C; right panel) at 12 hours (29.3% ± 5.8%, P < .01) compared to Ecoli vehicle-treated mice. Of importance in Figure 4C (right panel), the numbers of BM neutrophils remained nearly the same as at time 0.
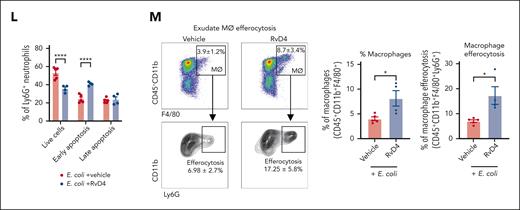
RvD4 stops neutrophil emergency deployment and accelerates the resolution of infection. Mice inoculated with E coli (105 CFU, IP) were also administered (IV) with RvD4 (100 ng per mouse) or vehicle alone (0.01% v/v ethanol in saline). Exudates, whole blood, and BM cells were collected at 0, 12, and 72 hours. (A) Schematic of the experimental design and sample collection. (B) CyTOF: UMAP of the infectious exudate, whole blood, and the BM, labeled with 17 immune populations. (C) Number of neutrophils in exudate, whole blood, and the BM. (D) Number of LSKs and GMPs in whole blood and the BM. (E) UMAP of neutrophil populations and progenitors in the BM, whole blood, and exudate. (F) Neutrophil lineage trajectory: diffusion map. LSKs, GMPs, PN (preneutrophils), IN (immature neutrophils), MN (mature neutrophils); and CN, circulating neutrophils. (G) RvD4 and E coli logFC change diffusion map calculated using edgeR with diffcyt. Only statistically significant populations are colored (P < .05) and adjusted using a Benjamini-Hochberg correction. Results in panels C-D are mean ± SEM; n = 3 samples biologically independent per time point. RvD4 vs E coli + vehicle; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001 using 1-way ANOVA with Tukey multiple comparison test. (H) Exudate: E coli bacterial titers (300 μL lavage) at 12 hours. Results are expressed as mean ± SEM; n = 8 or 9 samples per condition. RvD4 vs E coli + vehicle, ∗∗P < .01 using 2-tailed t test. (I-J) Exudate: in vivo phagocytosis. Intracellular E coli levels were determined in neutrophils (CD45+F4/80−Ly6C−Ly6G+), monocytes (CD45+F4/80−Ly6G−Ly6C+), and macrophages (CD45+Ly6G−Ly6C−F4/80+). MFI, mean fluorescence intensity. (I) Representative histograms. (J) Quantification of MFI of intracellular E coli levels. Results are expressed as mean ± SEM; n = 4 samples; RvD4 vs E coli + vehicle, ∗P < .05 using 2-way ANOVA with Bonferroni multiple comparisons test. (K-L) Flow cytometry: exudate neutrophil apoptosis (CD45+Ly6G+annexinV+) at 12 hours. (K) Representative dot plots and (L) quantification of neutrophil apoptosis. Results are expressed as mean ± SEM; n = 4 or 5 samples. RvD4 vs E coli + vehicle, ∗∗∗∗P < .0001 using 2-way ANOVA with Bonferroni multiple comparisons test. (M) Flow cytometry: exudate in vivo macrophage efferocytosis (CD45+CD11b+F4/80+Ly6G+) at 12 hours. Results are expressed as mean ± SEM; n = 4 or 5 samples per condition. RvD4 vs E coli + vehicle, ∗P < .05 using 2-tailed t test.
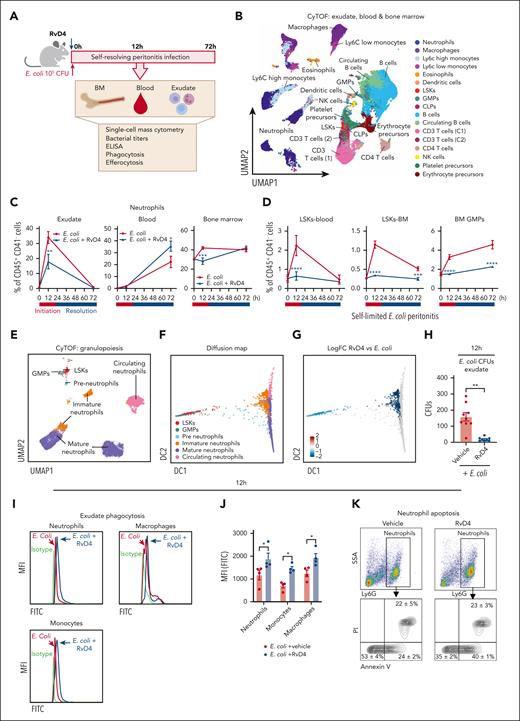
At 12 hours, RvD4 also resulted in a reduction in the number of circulating LSKs, BM-LSKs, and BM-GMPs by ∼89%, ∼61%, and ∼53%, respectively, compared mice with peritonitis treated with vehicle only (Figure 4D). Notably, the percentage of circulating LSKs, BM-LSKs, and BM-GMPs in RvD4-treated mice remained essentially the same as that at time 0 (Figure 4D). We questioned whether RvD4 affects the lineage trajectory of granulocyte multipotent progenitors and neutrophils. Neutrophils and their progenitors were further analyzed using UMAP-FlowSOM during the time course of peritonitis. We identified 5 clusters: LSKs, GMPs, preneutrophils, immature neutrophils, and mature neutrophils that originated from the BM and represented differentiating neutrophils, whereas 1 cluster of circulating neutrophils was mainly found in the peripheral blood and exudates (Figure 4E) (supplemental Figure 3C, compartment-specific clustering). Next, we used the UMAP from Figure 4E and performed nonlinear dimensionality reduction using diffusion maps.23 Diffusion maps allow granulocytes to be computationally organized along their lineage trajectory to identify cluster-specific changes during the time course of infections. In Figure 4F, the neutrophil progenitors LSKs, GMPs, and preneutrophils resided at 1 end of the diffusion map, whereas immature, mature, and circulating neutrophils were at the opposite end, organizing the cells based on their known lineage trajectory and differentiation. To assess whether RvD4 initiated changes in granulocyte lineage trajectory during infections, we performed differential abundance analysis using diffcyt22 from the diffusion map in Figure 4F. At 12 hours, the peak of inflammation, RvD4-treated mice had a log fold-change downregulation in granulocyte lineage trajectory in LSKs, GMPs, preneutrophil, and immature neutrophils. By contrast, RvD4 did not change the lineage trajectory of the terminally differentiated cells, mature neutrophils and circulating neutrophils (Figure 4G).
RvD4 stimulates bacterial clearance, and regulates granulopoiesis and deployment
Given that RvD4 limits neutrophil infiltration into the exudate and prevents excessive BM neutrophil deployment (Figures 3 and 4C), we set out to determine the impact of RvD4 on bacterial clearance. In Figure 4H, RvD4 resulted in a marked reduction of ∼75% in bacterial titers in exudates at 12 hours compared with E coli vehicle–treated mice. Bacterial titer photographs of exudates are shown in supplemental Figure 3D. Next, we determined whether RvD4 directly affects bacterial containment and phagocytosis in vivo and in vitro. Peritoneal exudates were collected at 12 hours and assayed for intracellular E coli ingestion by neutrophils, monocytes, and macrophages using flow cytometry. RvD4 resulted in a statistically significant enhanced containment of E coli in vivo by neutrophils (∼58%, P < .05), monocytes (∼48%, P < .05), and macrophages (∼35%, P < .05) when compared with mice treated with E coli and only vehicle (Figure 4I-J). We also isolated naïve peritoneal macrophages for in vitro phagocytosis experiments using fluorescently labeled live E coli incubated with RvD4 (0.1-10 nM) or only vehicle (supplemental Figure 3E-F). RvD4 at 1 nM resulted in the highest increase in phagocytosis, at ∼45% (P < .05), compared with E coli treatment alone. Fluorescent images of macrophage phagocytosis are shown in supplemental Figure 3G.
Because neutrophil apoptosis is one of the signals that initiates the resolution phase of inflammation, we determined whether RvD4 alters neutrophil viability. Peritoneal exudates were collected at 12 hours after infection, and neutrophil apoptosis was measured by flow cytometry. RvD4 resulted in increased neutrophil apoptosis (CD45+Ly6G+AnnexinV+PI−) by ∼40% (P < .001) when compared with mice treated with E coli and vehicle only (Figure 4K-L). Given that neutrophil clearance is a fundamental hallmark of resolution, we evaluated whether RvD4 initiates macrophage efferocytosis of apoptotic neutrophils in vivo. RvD4-treated mice had increases in the percentage of macrophages with ingested neutrophils (CD45+CD11b+F4/80+Ly6G+) at 12 hours (Figure 4M). Notably, RvD4 also resulted in a doubled percentage of macrophages in exudates (CD45+CD11b+F4/80+) compared with mice treated with E coli and vehicle only (Figure 4M).
We monitored the levels of granulocyte colony-stimulating factor (G-CSF), CXCL1(KC), and CXCL12 at the peak of neutrophilic infiltration at 12 hours (eg, Figure 1A-C), because these molecules are known to control BM neutrophil deployment.2 RvD4 resulted in decreased amounts of G-CSF in both the circulation and BM (Figure 5A). RvD4 also resulted in decreased levels of CXCL1(KC) in both the circulation and infectious exudates compared with levels in mice treated with E coli and vehicle only (Figure 5B). Amounts of circulating CXCL12 decreased in RvD4-treated mice, whereas the amounts in the BM were higher compared with those in mice treated with vehicle only (Figure 5C). We assessed direct actions of RvD4 on PMN in whole blood. Naïve mice were injected IV with leukotriene B4 (LTB4), an endogenous proinflammatory lipid mediator and a potent neutrophil chemoattractant.24 Within 5 minutes, LTB4 stimulated a rapid increase in PMN numbers in whole blood that reached a maximum at 10 minutes (Figure 5D). PMN numbers returned to basal values by ∼60 minutes. RvD4 alone did not evoke PMN increases in the whole blood. Next, we determined whether RvD4 could stop LTB4-induced PMN mobilization. Here, mice were systemically given either RvD4 or vehicle, 10 minutes before IV administration of LTB4. RvD4 blocked LTB4-initiated PMN mobilization by ∼60% at 10 minutes, which was sustained.

RvD4 regulates neutrophil infiltration in a dose-dependent manner. (A-C) Mice inoculated with E coli (105 CFU, IP) were also (IV) administered RvD4 (100 ng per mouse) or vehicle alone (0.01% v/v ethanol in saline). Exudates, whole blood, and BM cells were collected at 12 hours. Levels of G-CSF, CXCL1, and CXCL12 in the serum, BM, and exudate. Results are expressed as mean ± SEM; n = 3 to 5 samples per condition. RvD4 vs E coli + vehicle, ∗P < .05, ∗∗P < .01, ∗∗∗P < .001 using 2-tailed t test. (D) LTB4 whole-blood neutrophil mobilization. Naïve mice were IV administered LTB4 (red line), RvD4 (dark blue line), combination of RvD4 + LTB4 (purple), or vehicle only (0.01% ethanol v/v in saline). Neutrophils were enumerated by light microscopy and differential count. Results are expressed as mean ± SEM; n = 6 mice per condition. Time 0 minute vs 5, 10, 15, 20, 30, 40, and 60 minutes, respectively; ∗P < .05, ∗∗∗ P < .001, ∗∗∗∗P < .0001. RvD4 + LTB4 vs LTB4; ###P < .001, using 2-way ANOVA with Bonferroni multiple comparisons test. (E-G) Mice were inoculated with E coli (105 CFU, IP) and administered (IV) RvD4 at concentrations of 1, 10, or 100 ng per mouse, or vehicle only (0.01% v/v ethanol in saline). Exudates and whole blood were collected at 12 hours. (E) Cell number of neutrophils (CD45+F4/80−Ly6C−Ly6G+) in peritoneal exudates; (F) levels of CXCL1 in exudate; (G) levels of G-CSF and CXCL12 in the serum. (E-G) Results are expressed as mean ± SEM; n = 7 (RvD4, 1 ng per mouse), n = 8 (RvD4, 10 ng per mouse), n = 4 (RvD4, 100 ng per mouse), or n = 9 (E coli + vehicle). RvD4 vs E coli + vehicle; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001 with 2-way ANOVA with Bonferroni multiple comparisons test.
RvD4 regulates neutrophil infiltration in a dose-dependent manner. (A-C) Mice inoculated with E coli (105 CFU, IP) were also (IV) administered RvD4 (100 ng per mouse) or vehicle alone (0.01% v/v ethanol in saline). Exudates, whole blood, and BM cells were collected at 12 hours. Levels of G-CSF, CXCL1, and CXCL12 in the serum, BM, and exudate. Results are expressed as mean ± SEM; n = 3 to 5 samples per condition. RvD4 vs E coli + vehicle, ∗P < .05, ∗∗P < .01, ∗∗∗P < .001 using 2-tailed t test. (D) LTB4 whole-blood neutrophil mobilization. Naïve mice were IV administered LTB4 (red line), RvD4 (dark blue line), combination of RvD4 + LTB4 (purple), or vehicle only (0.01% ethanol v/v in saline). Neutrophils were enumerated by light microscopy and differential count. Results are expressed as mean ± SEM; n = 6 mice per condition. Time 0 minute vs 5, 10, 15, 20, 30, 40, and 60 minutes, respectively; ∗P < .05, ∗∗∗ P < .001, ∗∗∗∗P < .0001. RvD4 + LTB4 vs LTB4; ###P < .001, using 2-way ANOVA with Bonferroni multiple comparisons test. (E-G) Mice were inoculated with E coli (105 CFU, IP) and administered (IV) RvD4 at concentrations of 1, 10, or 100 ng per mouse, or vehicle only (0.01% v/v ethanol in saline). Exudates and whole blood were collected at 12 hours. (E) Cell number of neutrophils (CD45+F4/80−Ly6C−Ly6G+) in peritoneal exudates; (F) levels of CXCL1 in exudate; (G) levels of G-CSF and CXCL12 in the serum. (E-G) Results are expressed as mean ± SEM; n = 7 (RvD4, 1 ng per mouse), n = 8 (RvD4, 10 ng per mouse), n = 4 (RvD4, 100 ng per mouse), or n = 9 (E coli + vehicle). RvD4 vs E coli + vehicle; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001 with 2-way ANOVA with Bonferroni multiple comparisons test.
Next, we determined whether RvD4 at lower concentrations could also control excessive neutrophil infiltration during infection. RvD4 was administered (IV) at concentrations of 1 ng per mouse, 10 ng per mouse, 100 ng per mouse, or vehicle, to mice with self-limited bacterial infections. Peritoneal exudates and whole blood were collected at 12 hours (ie, peak of inflammation, Figure 1A). RvD4 administration significantly reduced the number of neutrophils infiltrating the peritoneal cavity at concentrations of 1 ng per mouse (P < .001), 10 ng per mouse (P < .01), and 100 ng per mouse (P < .01) (Figure 5E). ANOVA showed that there were no statistically significant differences (P = .98) among all 3 concentrations of RvD4, indicating that they essentially had the same potency in vivo (Figure 5E). RvD4 treatment resulted in a statistically significant decrease in the levels of CXCL1(KC) in the exudate at all 3 concentrations (Figure 5F). We determined the levels of G-CSF and CXCL12 in the serum from these mice. RvD4 resulted in a decrease in the levels of both G-CSF and CXCL12 in circulation at 12 hours (Figure 5G).
RvD4 accelerates macrophage efferocytosis of aged BM neutrophils
We questioned whether BM macrophages can also engage in Rv-regulated efferocytosis. BM cells were collected from mice with bacterial peritonitis from RvD4 treated mice and vehicle alone. BM macrophages were identified as CD169+F4/80+CD11b+VCAM+ and ingested neutrophils based on their intracellular expression of Ly6G+ (supplemental Figure 4A) by flow cytometry. Mice with peritonitis treated with RvD4 showed an increased percentage of CD169+-macrophage efferocytosis of BM neutrophils, for example, at 72 hours (53.1% ± 11.5%; P < .05) compared with that in mice treated with vehicle only (19.4% ± 4.3%) (Figure 6A). To investigate RvD4’s direct actions on BM macrophages, naïve BM-derived macrophages (BMDM) were incubated with RvD4 (0.01-100 nM) or vehicle before exposure of aged BM neutrophils. RvD4 resulted in increased efferocytosis of aged neutrophils, which was dose-dependent, with an estimated 50% effective concentration of ∼0.1 nM (Figure 6B). We also used confocal microscopy to visualize BMDM efferocytosis of aged neutrophils in the presence of either RvD4 (1 nM) or vehicle only. Confocal microscopy images and 3-dimensional rendering movies illustrated that BMDM treated with RvD4 substantially increased the uptake of neutrophils compared with cells exposed to vehicle only (Figures 6C; supplemental Figure 4B-C; supplemental Videos 1 [RvD4] and 2 [vehicle]).
![RvD4 increases the CD169+ macrophage efferocytosis of neutrophils in the BM and regulates lineage differentiation. Mice were inoculated with E coli (105 CFU, IP) and received RvD4 (100 ng per mouse, IV) or vehicle only (0.01% v/v ethanol in saline). At 12 and 72 hours after inoculation, BM cells were collected. (A) Flow cytometry for efferocytosis of BM neutrophils by BM macrophages in vivo. Results are mean ± SEM; n = 3 mice per time point; ∗P < .05, vs E coli + vehicle. Statistical analysis was carried out using 1-way ANOVA with Tukey multiple comparison test. (B) Flow cytometry BM macrophage efferocytosis of aged neutrophils (see “Methods”). Dose response: RvD4-induced percent increase in BMDM efferocytosis of aged BM neutrophils relative to that in vehicle-treated cells (solid black line). The 50% effective concentration (EC50) was estimated using nonlinear regression (dashed green line) with log (RvD4) vs response (3 parameters). Results are expressed as mean ± SEM; n = 5 (aged BM neutrophils); ∗P < .05, ∗∗∗P < .001, ∗∗∗∗P < .0001 when compared with vehicle (as control) using 1-way ANOVA with Bonferroni multiple comparison test. (C) Confocal microscopy of BMDM efferocytosis of aged BM neutrophils. The scale bars represent 20 μm (see supplemental Videos 1 [RvD4] and 2 [vehicle] showing 3D reconstruction). The white arrows denote ingested neutrophils by a macrophage. Yellow arrow denotes (supplemental Video 1, RvD4) the #3 neutrophil ingested by a macrophage. Representative of n = 4 mice. (D) CyTOF: UMAP of BM leukocytes single-cell lineage trajectory. (E) CyTOF: BM violin scatter plot of LSKs single-cell trajectory analysis at 12 hours after E coli inoculation. (F) Densities pseudotime plot trajectories. The density plots reflect differences in BM cell densities between RvD4 + E coli vs E coli plus vehicle treatments, at 12 hours. (G) BM myeloid CFUs: M, monocytes; G, granulocyte; and GM, macrophage/granulocyte at 12 hours after E coli inoculation or in naïve control mice. Results are mean ± SEM; n = 5 (naïve) or n = 9 samples (E coli + RvD4, or E coli + vehicle) per time point. E coli + RvD4 vs E coli + vehicle or vs naïve; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. Statistical analysis was carried out using 2-way ANOVA with Bonferroni multiple comparisons test.](/view-large/figure/11799728/BLOOD_BLD-2022-019145-gr6.jpg)
RvD4 increases the CD169+ macrophage efferocytosis of neutrophils in the BM and regulates lineage differentiation. Mice were inoculated with E coli (105 CFU, IP) and received RvD4 (100 ng per mouse, IV) or vehicle only (0.01% v/v ethanol in saline). At 12 and 72 hours after inoculation, BM cells were collected. (A) Flow cytometry for efferocytosis of BM neutrophils by BM macrophages in vivo. Results are mean ± SEM; n = 3 mice per time point; ∗P < .05, vs E coli + vehicle. Statistical analysis was carried out using 1-way ANOVA with Tukey multiple comparison test. (B) Flow cytometry BM macrophage efferocytosis of aged neutrophils (see “Methods”). Dose response: RvD4-induced percent increase in BMDM efferocytosis of aged BM neutrophils relative to that in vehicle-treated cells (solid black line). The 50% effective concentration (EC50) was estimated using nonlinear regression (dashed green line) with log (RvD4) vs response (3 parameters). Results are expressed as mean ± SEM; n = 5 (aged BM neutrophils); ∗P < .05, ∗∗∗P < .001, ∗∗∗∗P < .0001 when compared with vehicle (as control) using 1-way ANOVA with Bonferroni multiple comparison test. (C) Confocal microscopy of BMDM efferocytosis of aged BM neutrophils. The scale bars represent 20 μm (see supplemental Videos 1 [RvD4] and 2 [vehicle] showing 3D reconstruction). The white arrows denote ingested neutrophils by a macrophage. Yellow arrow denotes (supplemental Video 1, RvD4) the #3 neutrophil ingested by a macrophage. Representative of n = 4 mice. (D) CyTOF: UMAP of BM leukocytes single-cell lineage trajectory. (E) CyTOF: BM violin scatter plot of LSKs single-cell trajectory analysis at 12 hours after E coli inoculation. (F) Densities pseudotime plot trajectories. The density plots reflect differences in BM cell densities between RvD4 + E coli vs E coli plus vehicle treatments, at 12 hours. (G) BM myeloid CFUs: M, monocytes; G, granulocyte; and GM, macrophage/granulocyte at 12 hours after E coli inoculation or in naïve control mice. Results are mean ± SEM; n = 5 (naïve) or n = 9 samples (E coli + RvD4, or E coli + vehicle) per time point. E coli + RvD4 vs E coli + vehicle or vs naïve; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. Statistical analysis was carried out using 2-way ANOVA with Bonferroni multiple comparisons test.
RvD4 increases the CD169+ macrophage efferocytosis of neutrophils in the BM and regulates lineage differentiation. Mice were inoculated with E coli (105 CFU, IP) and received RvD4 (100 ng per mouse, IV) or vehicle only (0.01% v/v ethanol in saline). At 12 and 72 hours after inoculation, BM cells were collected. (A) Flow cytometry for efferocytosis of BM neutrophils by BM macrophages in vivo. Results are mean ± SEM; n = 3 mice per time point; ∗P < .05, vs E coli + vehicle. Statistical analysis was carried out using 1-way ANOVA with Tukey multiple comparison test. (B) Flow cytometry BM macrophage efferocytosis of aged neutrophils (see “Methods”). Dose response: RvD4-induced percent increase in BMDM efferocytosis of aged BM neutrophils relative to that in vehicle-treated cells (solid black line). The 50% effective concentration (EC50) was estimated using nonlinear regression (dashed green line) with log (RvD4) vs response (3 parameters). Results are expressed as mean ± SEM; n = 5 (aged BM neutrophils); ∗P < .05, ∗∗∗P < .001, ∗∗∗∗P < .0001 when compared with vehicle (as control) using 1-way ANOVA with Bonferroni multiple comparison test. (C) Confocal microscopy of BMDM efferocytosis of aged BM neutrophils. The scale bars represent 20 μm (see supplemental Videos 1 [RvD4] and 2 [vehicle] showing 3D reconstruction). The white arrows denote ingested neutrophils by a macrophage. Yellow arrow denotes (supplemental Video 1, RvD4) the #3 neutrophil ingested by a macrophage. Representative of n = 4 mice. (D) CyTOF: UMAP of BM leukocytes single-cell lineage trajectory. (E) CyTOF: BM violin scatter plot of LSKs single-cell trajectory analysis at 12 hours after E coli inoculation. (F) Densities pseudotime plot trajectories. The density plots reflect differences in BM cell densities between RvD4 + E coli vs E coli plus vehicle treatments, at 12 hours. (G) BM myeloid CFUs: M, monocytes; G, granulocyte; and GM, macrophage/granulocyte at 12 hours after E coli inoculation or in naïve control mice. Results are mean ± SEM; n = 5 (naïve) or n = 9 samples (E coli + RvD4, or E coli + vehicle) per time point. E coli + RvD4 vs E coli + vehicle or vs naïve; ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. Statistical analysis was carried out using 2-way ANOVA with Bonferroni multiple comparisons test.
RvD4 signals BM granulocyte progenitors: cell trajectories
HSPCs are responsible for the maintenance of cellular components of the peripheral blood, including neutrophils.2 The fate decisions of HSPCs during infection can have a significant impact on the host to either respond or delay the resolution phase of inflammation. We investigated whether RvD4 affects BM HSPC (LSKs) fate decision during self-limited infections. Therefore, we performed pseudotime analysis (Figures 6D-F) that allows the measurements of single-cell changes during HSPC (LSK) fate differentiation by temporally organizing cells along lineage trajectories using the algorithm Slingshot.25 BM cell trajectories, at 12 hours, were calculated from the UMAP in Figure 4B containing BM annotated populations. BM lineage trajectories were rooted from LSKs, because this population comprises hematopoietic stem cells and multipotent progenitors.4 As shown in Figure 6D, Slingshot identified 2 distinct lineage trajectories in the BM: (1) myeloid (LSKs, GMPs, eosinophils, monocytes [Ly6Chigh and Ly6Clow], macrophages, and neutrophils) and (2) lymphoid (LSKs, CLPs, and CD3+, CD4+, natural killer, and B cells). Along this modeled trajectory, BM cells from RvD4-treated mice (blue) had a decreased (lineage 1) myeloid lineage trajectory when compared with E coli + vehicle–treated mice at 12 hours (red) (Figure 6E-F). Density pseudotime differential progression analysis revealed that RvD4 favors LSK differentiation toward the lymphoid lineage (lineage 2), instead of the myeloid lineage (Figure 6F, left panel), with an increase in the density plot toward the end of the pseudotime trajectory (Figure 6F, right panel).
Given that RvD4 regulates granulocyte lineage differentiation during infections (Figure 6D-F), we determined whether RvD4 regulates myeloid clonogenic potential (Figure 6G) using CFU assay with BM cells of mice with peritonitis at 12 hours. RvD4-treated mice had decreased numbers of BM CFU-G and CFU-GM compared with that in the BM of vehicle-treated mice (Figure 6G). The numbers of CFU-M appeared not to change between RvD4-treated and E coli + vehicle–treated groups (Figure 6G). Notably, there was no statistical difference in CFU-G and CFU-GM between E coli + RvD4–treated mice and CFUs from naïve mice.
Intracellular signals activated by RvD4 in human and mouse cells
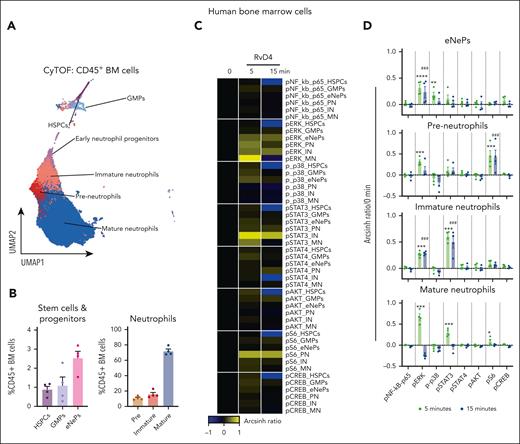
Because RvD4 prevented excessive BM neutrophil deployment and regulated the differentiation of granulocytes during distal infections (Figures 4 and 6), we investigated whether RvD4 initiated intracellular signaling within the human BM to interrogate pathways involved in granulopoiesis (ie, MAPK, PI3K/AKT, and JAK/STAT18) using single-cell mass cytometry. As shown in Figure 7, BM aspirates were incubated with RvD4 (10 nM) for 0, 5, or 15 min and then stained with a panel of antibodies specific for 28 cell surface proteins and 7 intracellular phosphorylated proteins. Human BM CD45+ cells (supplemental Figure 5A) were analyzed using UMAP-FlowSOM21 to identify granulocytes and their progenitors (Figure 7A). Cell surface markers known to be present in human BM neutrophils and granulocyte progenitors26 are listed in supplemental Table 3 and supplemental Figure 5B-C. Figure 7B demonstrates that neutrophil populations comprise ∼85% of human BM leukocytes. Within these populations, RvD4-stimulated intracellular signaling responses (changes in phosphorylation state) are summarized in a heat map in Figure 7C. Specifically, in preneutrophils, immature neutrophils, and mature neutrophils, RvD4 resulted in a statistically significant (P < .05) increase in phosphorylation of S6 protein and extracellular signal–regulated kinases 1 and 2 (pERK1/2), and was an activator of transcription (STAT) family members STAT3 by 5 minutes, which remained until 15 minutes (Figure 7D).
RvD4 stimulates phosphorylation during human neutrophil differentiation. Human BM aspirates were incubated for 5 and 15 minutes at 37°C with RvD4 (10 nM) or vehicle (0.01% v/v ethanol). CyTOF was carried out using a panel of antibodies targeting intracellular signaling phosphoprotein. (A) BM UMAP granulocyte populations and progenitors. (B) Number of neutrophil populations and progenitors identified in the BM. (C) Heatmap of the mean fold changes in the abundance of the intracellular phosphoproteins in BM cell populations of RvD4-treated aspirates relative to those in aspirates treated with vehicle only (median intensity arcsinh ratio). CREB, cAMP-response element binding protein. (D) Changes in the abundance of the intracellular phosphoproteins in BM cell populations. Results in panel D are expressed as mean ± SEM of n = 4 individual BM donors; 0 vs 5 minutes (RvD4): ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001, and 0 vs 15 minutes (RvD4): ###P < .001 by 1-way ANOVA with Bonferroni multiple comparison test. (E) Flow cytometry of human peripheral blood neutrophils (CD45+CD14−CD15+) phagocytosis of Bac-light Green–labeled E coli. (Top) Representative dot plot identifying human neutrophils. (Bottom) Representative histograms of geometric MFIs of Bac-light Green–labeled E coli in neutrophils. (F) Dose response: RvD4-induced percent increase in neutrophil phagocytosis of Bac-light Green–labeled E coli relative to that in vehicle-treated samples. ∗P < .05, ∗∗∗∗P < .0001 when compared with vehicle control. Results are expressed as mean ± SEM, n = 4 healthy human donors. ∗P < .05, ∗∗∗∗P < .0001 when compared with vehicle control. EC50 was estimated using nonlinear regression (dashed line) with log (agonist) vs response (3 parameters). (G) Flow cytometry: calcium influx in human peripheral blood neutrophils incubated with 10 nM of either LTB4, RvD4, or RvD1. Results from n = 4 healthy human donors. (H-I) Flow cytometry: heat maps of phosphorylated signaling ERK1/2 (H) and STAT3 (I) at 0, 1, 5, and 15 minutes after incubation with RvD1 (10 nM) or RvD4 (10 nM) in Fpr2−/− (ALX receptor–deficient mice) and Fpr2flox/flox. Phosphorylation levels were calculated as the difference between the geometric mean signal intensity in RvD1- or RvD4-treated BM neutrophils (at 0, 1, 5, and 15 minutes) and the geometric mean signal intensity in vehicle-treated BM neutrophils at 0 minutes. Results are expressed as mean ± SEM, n = 3 mice from each group; 0 minute vs 1, 5, or 15 minutes, respectively; ∗P < .05, ∗∗P < .01.
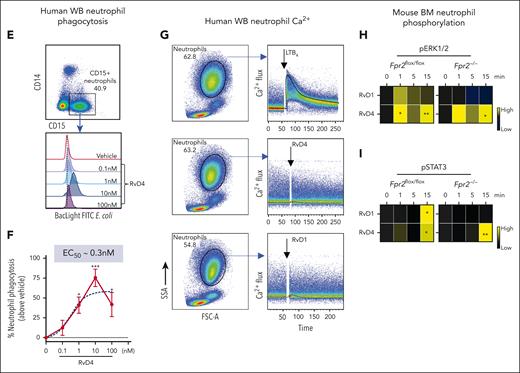
Given that RvD4 increased neutrophil phagocytosis in mouse exudates, we determined whether RvD4 enhances phagocytosis of live E coli in human whole-blood neutrophils (CD45+CD14−CD15+). RvD4 resulted in increased neutrophil phagocytosis in a dose-dependent manner, with the highest response at 10 nM (P < .001) compared with the vehicle controls, giving an estimated 50% effective concentration of ∼0.3 nM (Figure 7E-F). Because RvD4 induces phosphorylation in human BM granulocytes, we assessed whether RvD4 elicits intracellular Ca2+ release as a second messenger after receptor activation. In direct comparisonswith LTB4 (10 nM), neither RvD4 at 10 nM nor RvD1 mobilized intracellular Ca2+ in peripheral human whole-blood neutrophils (CD45+CD14−CD15+) (Figure 7G).
Rvs initiated their protective actions in leukocytes via activation of specific G protein–coupled receptors (GPCR),27 for example, RvD1 activates the Fpr2, ALX receptor in mice.28,29 Next, we investigated RvD4 receptor–initiated intracellular signaling in naïve BM cells from Fpr2−/− (ALX receptor–deficient mice) and Fpr2flox/flox mice. BM cells were incubated with RvD4 (10 nM), RvD1 (10 nM), or vehicle for 0 to 15 minutes. BM neutrophils were identified as (CD45+Ly6C−Ly6G+) (supplemental Figure 5D) using flow cytometry. RvD1 and RvD4 resulted in a time-dependent increase in phosphorylation of ERK1/2 and STAT3 as early as 1 minute (Figure 7H-I). In BM neutrophils from Fpr2-deficient mice, RvD1 phosphorylation of these 2 proteins was abolished. In contrast, RvD4-initiated phosphorylation of ERK1/2 and STAT3 in Fpr2−/− BM neutrophils persisted (Figure 7I), thus indicating that RvD4-initiated phosphorylation is ALX/FPR2 receptor independent.
Discussion
Deployment of neutrophils from the BM is a critical process in host defense, homeostasis, and during the acute inflammatory response.1,2 In this manuscript, we report that distal infections signal the BM via lipid mediators. Specifically, D-series Rvs, RvD1 and RvD4, are present in the BM, in which they have cell type–specific actions on BM myeloid cells. RvD4 proved to potently limit neutrophil infiltration to sites of infection and stop excessive neutrophil departure from the BM. Recent analyses of >31 245 patients demonstrate that excess uncontrolled inflammation is a strong predictor of cardiovascular death and all causes of death.30 These results emphasize the urgent need to harness natural mechanisms to resolve inflammation.
Rvs are produced in the resolution phase of peritonitis3,31 and released to the blood compartment in which they activate phagocytosis of E coli.3 Herein, we uncovered that RvD4 stops excessive neutrophil deployment. Results in Figures 1 and 2 demonstrate that self-limited bacterial peritonitis evokes neutrophil deployment and emergency granulopoiesis. These actions are accompanied by time-dependent changes in BM RvD1 and BM RvD4. Both RvD1 and RvD4 were recently identified in humans.32-35 Although human phagocytes, that is, neutrophils and macrophages, appear to be one of the main sources of RvD4,17 using LC-MS/MS-based identification, it has also been identified in other human tissues, for example, in the cerebrospinal fluid of patients with Alzheimer disease,35 in the peripheral blood of women with coronary microvascular dysfunction,32 in the plasma of adolescents,33 and in human airway epithelial cells.34 In mice, RvD1 and RvD4 are also present in the spleen, lungs, colon, and brain.36 In mouse BM, we also identified RvD5, 17-HDHA, and 18-HEPE, suggesting that proresolving mediators may have a role(s) in BM functions during infection. For example, RvD531 potently stimulates the killing and clearance of bacteria. Each pathway marker, 17-HDHA and 18-HEPE, is proven to be potent in reducing pain37 and is cardioprotective,38 respectively.
A systematic analysis of local mediators produced in the resolution phase of self-resolving acute inflammatory response(s) gave rise to Rvs and other SPMs that actively shorten the resolution interval.39 Each SPM total organic synthesis confirmed their potent agonist activities in preventing myeloid-mediated collated tissue damage and established their complete stereochemical assignments.40 Of the proresolving mediators, RvD4 appears later in the resolution phase of acute murine infections.15,16 Similar to most autocoids, lipid mediators are rapidly inactivated in their local milieu.39 In the case of RvD4, which stimulates phagocytosis of E coli11 and is organ protective11 after receptor activation, RvD4 is enzymatically inactivated via local conversion to the 17-oxo RvD4 in mouse BM and human BM11,41 (supplemental Figure 6). During the resolution of infectious inflammation, RvD4 regulates neutrophil deployment and RvD4 concentrations diminish in the BM (Figure 2A). These results indicate that RvD4 is a multifunctional signal. Thus, to assess the amounts of RvD4 produced, in further studies, it will be useful to monitor both 17-oxo-RvD4 and 15,16-dihydro-RvD4,11 metabolites that can be summed with RvD4 in order to quantitatively access RvD4 local production.
By definition, proresolving mediators limit further neutrophil recruitment to sites of inflammation, clearing apoptotic neutrophils and microbial infections.3 These are the phagocyte hallmarks of active resolution of inflammation.3 Each of the stereochemically defined SPM accelerates resolution.3,40 Results from direct comparisons and add-back experiments proved that these proresolving functions were confirmed for RvD1 and RvD4 in this study (Figure 3B). Here, we establish that RvD4 controls excessive (1) neutrophil deployment, (2) granulopoiesis, and (3) HSPC differentiation (as shown in Figures 3, 4, and 6). Along these lines, RvD1 also increases the numbers of Ly6Clow reparative monocytes (Figures 3D; supplemental Figure 2E). Together these results emphasize the unique structure-function relationship(s) of each Rv’s stereochemical structure, namely that RvD1 (7S,8R,17S-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid)14 activates the ALX/FPR2, the RvD1 receptor in mice,28 and that of RvD4 (4S,5R,17S-trihydroxydocosa-6E,8E,10Z,13Z,15E,19Z hexaenoic acid).11
RvD1 diminishes tumor burden by increasing the infiltration of antitumor monocytes.42 A metabolically stable epimer of RvD1, aspirin-triggered RvD1 (AT-RvD1), targets accumulation of proangiogenic neutrophils, which increases early vessel and wound remodeling.43 With human neutrophils, AT-RvD1 accelerates their apoptosis and phagocytosis.44 RvD4 also protects mice from neutrophil-mediated collateral tissue damage in ischemia-reperfusion injury11 and limits the formation of neutrophil extracellular traps in vivo, reducing the impact of deep vein thrombosis.45,46 Results from these studies11,42,44-46 and the experiments presented herein demonstrate that RvD1 and RvD4 each regulate myeloid cells to accelerate resolution of inflammation.
Infections increase circulating neutrophils and immature neutrophils, a process known as “a shift to the left.”2 Results in Figure 4 demonstrate that RvD4 stopped deployment of both immature and mature neutrophils from the BM into circulation. RvD4 also enhanced neutrophil phagocytosis in vivo and reduced bacterial titers. RvD4, like the other Rvs,3 limits excess neutrophil infiltration yet increases their efficacy in bacterial clearance (Figure 4H-I; supplemental Figure 3E-F). Neutrophil recruitment to the site of infection is governed by cytokines and lipid mediators.1,24,39,47 LTB4, a potent neutrophil chemoattractant,24 is also a signal for deployment of neutrophils that is regulated by RvD4 (Figure 5D). RvD4 increased the number of early-apoptotic neutrophils in the exudates (Figure 4K-L), a noninflammatory cell death program.47 The proresolving mediator RvE1 also accelerates neutrophil apoptosis.48 Neutrophil clearance from inflamed tissue is a hallmark of resolution.3 In the BM, granulopoiesis is partly controlled by macrophage efferocytosis of aged neutrophils49 and their clearance during embryonic hematopoiesis.50 Our results, shown in Figure 6 and supplemental Figures 4 and 4M, provide evidence that RvD4 is a potent agonist of macrophage efferocytosis by increasing BM clearance of aged neutrophils and clearance of apoptotic neutrophils in the exudate. In addition to stimulating the removal of apoptotic cells by macrophages, SPMs (ie, LXA4) directly stimulates neutrophil retrotaxis,51 opening the possibility that SPMs can also activate reverse neutrophil transendothelial migration in vivo.52
HSPC proliferation and differentiation is increased during obesity, sleep disturbances, high-fat diets, glucose intolerance, physical inactivity, and psychosocial stress.53 Sleep disturbance is associated with increased differentiation of myeloid cells and peripheral inflammation.54 Disrupted sleep has recently been demonstrated to affect the amounts of Rvs in the peripheral blood.55 RvE1 modulates posttraumatic sleep during traumatic brain injury in mice by regulating peripheral inflammation.55 RvD4 regulates the differentiation of HSPCs to granulocyte progenitors in response to distal infections, as well as their mobilization into the circulation (Figures 4D,G and 6D-G). Other SPMs act directly on embryonic and tissue-specific stem cells. For example, NPD1/PD1 potently increases the differentiation of mouse embryonic stem cells to neural and cardiac cells.13 RvD156 and RvD257 regulate muscle stem cells and progenitors to accelerate muscle regeneration.
Specific Rvs activate intracellular signaling within human phagocytes.58 In human whole blood, RvD1 stimulates phosphorylation of ERK1/2 and CREB in neutrophils and CD14+ monocytes.58 Using single-cell CyTOF (Figure 7A-D), we uncovered that, in human BM, RvD4 stimulated phosphorylation of regulatory pathways involved in human granulopoiesis,18 for example, MAPK (pERK1/2, pp38), PI3K/AKT (S6 ribosomal protein), and JAK/STAT (STAT3). Phosphorylation of ERK1/2 and STAT3 are critical in granulocyte differentiation and regulation of neutrophil deployment during emergency granulopoiesis.59,60 Phosphorylation of STAT3 in human neutrophils is known to accelerate neutrophil apoptosis.61 SPMs (eg, RvE1, RvD1, RvD2, PD1, and maresin 1) exhibit potent stereoselective actions in human leukocytes via specific GPCRs.27 RvD1 reportedly enhanced revascularization and perfusion in mice during hindlimb ischemia that was ALX/FPR2 dependent.29 In human macrophages, RvD4 stimulated phagocytosis of E coli, which is cholera toxin sensitive11, suggesting a role for a GPCRs that remains to be identified. Results in Figures 7H,I indicate that RvD4 mediates phosphorylation of ERK1/2 and STAT3 that is independent of the ALX/FPR2 receptor and that did not mobilize intracellular calcium in human neutrophils.
In summary, these results indicate that RvD4 targets BM neutrophil deployment, differentiation, and tissue infiltration in response to distal infections. Moreover, our results suggest that targeting excessive neutrophil deployment and emergency granulopoiesis via SPMs (eg, RvD4) could be a therapeutic approach. Resolution agonists might be useful in pathologies linked to excessive neutrophil deployment and granulopoiesis (eg, trauma, sepsis, and myeloid disorders).
Acknowledgments
The authors thank M. H. Small for their expert assistance in manuscript preparation. The authors also thank Matthew Spite (CET&RI, Brigham and Women’s Hospital) for the BM cells from Fpr2−/− and Fpr2flox/flox mice, ALX receptor, Mélissa Simard (CET&RI, Brigham and Women's Hospital) for expert support on mouse sample collection, Nan Chiang (CET&RI, Brigham and Women's Hospital) for expert discussion, and the Dana-Farber–Harvard Medical School CyTOF Core for expert support on sample acquisition.
This work was supported, in part, by the National Institutes of Health (NIH), National Institute of General Medical Sciences (grants R35GM139430 and R01GM038765, C.N.S.), and NIH, National Heart, Lung, and Blood Institute (grant K99HL153673, S.L.). B.L. received support from Cayman Chemical (Ann Arbor, MI) for reagents.
Authorship
Contribution: S.L. and C.N.S. conceived and designed the experiments; S.L. performed and analyzed the CyTOF data, flow cytometry studies, confocal, animal studies, and LC-MS/MS results, as well as contributed to figure preparation; R.N. performed LC-MS/MS matching and analyzed LC-MS/MS results, as well as contributed to figure preparation; B.L. analyzed CyTOF data, as well as contributed to figure preparation; S.L. and C.N.S. wrote the manuscript; C.N.S. analyzed data, acquired funding, and supervised the project; and all authors reviewed and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Charles N. Serhan, Center for Experimental Therapeutics and Reperfusion Injury, Brigham and Women’s Hospital, 60 Fenwood Rd, Hale Building for Transformative Medicine 3-016, Boston, MA 02115; e-mail: cserhan@bwh.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal