

In this issue of Blood, Chen et al1 studied the metabolic adaptations of chronic lymphocytic leukemia (CLL) cells during their life cycle as they move in and out of the lymph nodes (LNs). They identified glutamine as an essential molecule and as an intermediary involved in resistance to venetoclax.
One of the fascinating aspects of CLL is the continuous and intense recirculation of leukemic cells through LNs. Via rapid and dynamic modulation of chemokine receptors and adhesion molecules, leukemic cells home to LNs where they receive growth and survival signals through the B-cell receptor (BCR) and other “accessory” molecules including CD40. Once in the LN, leukemic cells rapidly emerge and return to the circulation, thereby establishing a cycle that guarantees the clone survival and its relatively slow progression. The clinical success of Bruton tyrosine kinase (BTK) inhibitors is based on the interruption of this circuit, causing massive release of CLL cells from the LNs into the circulation, where they subsequently die, likely because of lack of positive signals.2,3
Studying CLL cells in the LN microenvironment is difficult because of the rarity of LN biopsies in patients with CLL. The authors addressed this problem by using 2 independent approaches. First, they studied leukemic cells purified from the blood of patients in the early stages of treatment with ibrutinib and used the specific markers CD5 and CXCR4 to identify recent LN emigrants. Second, they used normal circulating leukemic cells that were activated in vitro through the BCR and CD40, two major signaling molecules in the LN microenvironment. Analysis of glucose uptake and mitochondrial mass confirmed that these 2 cell populations were metabolically more active, suggesting that they had been correctly selected.4 By using metabolomic analyses through mass spectrometry complemented with gene expression studies, the authors show that glutamine is essential in determining the more energetic profile found in bona fide LN-resident leukemic cells. Glutamine uptake is greater in these cells than in their circulating counterparts and is the main substrate of the tricarboxylic acid cycle (TCA) (see figure). These findings are consistent with recent data underlining the importance of glutamine for tumor growth and promoting increased energy production, while at the same time protecting from the excessive production of reactive oxygen species (ROS).5 Although the exact molecular mechanisms remain unclear, glutamine metabolism may be driven by MYC, which in CLL, is activated through BCR signaling and relies on the upregulation of selective cell surface transporters, as documented in the study by Chen et al.
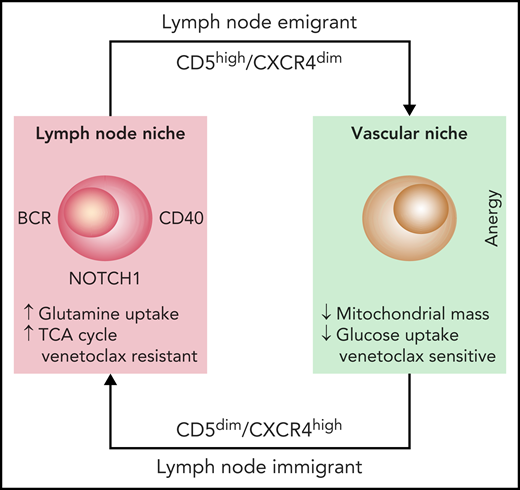
The diagram depicts the life cycle of CLL cells, constantly recirculating between the vascular and the LN niches. Once in the LN niche, CLL immigrants increase their uptake of glutamine to fuel the TCA cycle. This metabolic adaptation, triggered through microenvironmental interactions, favors venetoclax resistance, suggesting that targeting glutamine uptake may be beneficial.
The diagram depicts the life cycle of CLL cells, constantly recirculating between the vascular and the LN niches. Once in the LN niche, CLL immigrants increase their uptake of glutamine to fuel the TCA cycle. This metabolic adaptation, triggered through microenvironmental interactions, favors venetoclax resistance, suggesting that targeting glutamine uptake may be beneficial.
The idea that oncogenic signaling promotes metabolic adaptation is certainly not new, but our understanding of how CLL cells obtain energy is still incomplete. Previous studies using quiescent cells from the peripheral blood have generally concluded that CLL cells display heightened mitochondrial respiration, elevated ROS, and elevated antioxidant capacity.6 Those studies also addressed the issue of metabolic adaptation in and out of closed environments or in the presence of micro-environmental interactions. Interestingly, these studies identified a glycolytic switch occurring in the LNs, potentially because of the activation of the NOTCH1-MYC axis.7,8 The Chen et al study identifies, for the first time, the glutamine dependency of CLL cells that are receiving signals from the tumor niche. These findings may be actionable: the identification of a cell surface glutamine transporter that is upregulated after MYC activation may pave the way to novel therapies. These observations are of particular importance, given the emerging resistance to targeted therapies that rely on the activation of glutamine metabolism, as is the case for KRAS–driven lung cancers.9 Perhaps more relevant to CLL, other studies have shown synergy between drugs that target the glutamine pathway and venetoclax in models of acute leukemia and myeloma.10 Thus, designing drugs that interrupt metabolic circuits may overcome resistance to targeted therapies.
The study by Chen et al also has limitations: first and foremost, the models adopted by the researchers may be too simplistic in terms of rebuilding a niche in vitro or the models may be misleading by analyzing cells that have already been exposed to a targeted drug. Additional work is needed to determine the actual importance of these metabolic circuits. The second important limitation is that the authors have not segregated CLL cells according to their molecular lesions: this may be important for understanding CLL progression and eventual transformation into Richter syndrome. It will be interesting to know how specific genetic lesions directly impact the metabolic pathways of these cells. What remains a fascinating mystery is why CLL cells so rapidly leave the LNs instead of remaining in a nutrient-rich, favorable environment. What is that telling us about the cell of origin?
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal