

Key Points
Our findings identify a previously unrecognized role of the Gab2–MALT1 axis in IL-1β-induced thromboinflammation.
Pharmacological inhibition of MALT1 attenuates venous thrombosis induced by flow restriction.

Abstract
Deep vein thrombosis (DVT) is the third most common cause of cardiovascular mortality. Several studies suggest that DVT occurs at the intersection of dysregulated inflammation and coagulation upon activation of inflammasome and secretion of interleukin 1β (IL-1β) in restricted venous flow conditions. Our recent studies showed a signaling adapter protein, Gab2 (Grb2-associated binder 2), plays a crucial role in propagating inflammatory signaling triggered by IL-1β and other inflammatory mediators in endothelial cells. The present study shows that Gab2 facilitates the assembly of the CBM (CARMA3 [CARD recruited membrane-associated guanylate kinase protein 3]–BCL-10 [B-cell lymphoma 10]–MALT1 [mucosa-associated lymphoid tissue lymphoma translocation protein 1]) signalosome, which mediates the activation of Rho and NF-κB in endothelial cells. Gene silencing of Gab2 or MALT1, the effector signaling molecule in the CBM signalosome, or pharmacological inhibition of MALT1 with a specific inhibitor, mepazine, significantly reduced IL-1β–induced Rho-dependent exocytosis of P-selectin and von Willebrand factor (VWF) and the subsequent adhesion of neutrophils to endothelial cells. MALT1 inhibition also reduced IL-1β–induced NF-κB–dependent expression of tissue factor and vascular cell adhesion molecule 1. Consistent with the in vitro data, Gab2 deficiency or pharmacological inhibition of MALT1 suppressed the accumulation of monocytes and neutrophils at the injury site and attenuated venous thrombosis induced by the inferior vena cava ligation-induced stenosis or stasis in mice. Overall, our data reveal a previously unrecognized role of the Gab2–MALT1 axis in thromboinflammation. Targeting the Gab2–MALT1 axis with MALT1 inhibitors may become an effective strategy to treat DVT by suppressing thromboinflammation without inducing bleeding complications.
Introduction
Deep vein thrombosis (DVT) and pulmonary embolism, assigned together as venous thromboembolism, are among the leading causes of mortality globally.1-3 Unlike arterial thrombosis, which develops primarily from plaque rupture and platelet accumulation, venous thrombosis occurs without endothelial denudation and injury.4 Stasis of blood flow, intravascular vessel wall injury, and hypercoagulability are Virchow’s triad that contributes to thrombus development in DVT.5,6 The existing therapeutic strategies using anticoagulants are useful in deterring venous thrombosis; however, they are associated with an increased bleeding risk.7-9 At present, the molecular events contributing to DVT are not fully known.
The complex interplay among the coagulation system, endothelial cells, leukocytes, and interleukins (ILs) plays a crucial role in the pathogenesis of DVT.4,10,11 Consequently, venous thrombosis is tightly associated with thromboinflammation.12,13 It was recently shown that the proinflammatory cytokine IL-1β drives hypoxia and inflammation-driven hypercoagulability in DVT.14-16 Moreover, IL-1β activates endothelial cells and induces the expression of tissue factor (TF) and other prothrombotic genes that directly contribute to fibrin generation.17 The blockade of IL-1 receptor (IL-1R) signaling using monoclonal antibodies protects from leukocyte recruitment and thrombus development.16 However, treating inflammation by antagonists that block the IL-1R or other inflammatory receptors may prone patients to infections.18,19 Furthermore, they may also lead to the development of antidrug antibodies.20
Endothelial release of von Willebrand factor (VWF) and translocation of P-selectin to the cell surface that promote adhesion of platelets and neutrophils to the vessel wall play a crucial role in the thrombus development.21 In addition to VWF and P-selectin, the generation of neutrophil extracellular traps (NETs) provides additional scaffolds for the adhesion of platelets and coagulation factors.22,23 Degeneration of NETs by heparin or DNase1 treatment protects against DVT.24 Currently, most of the studies on the pathogenesis of DVT are focused on coagulation. The precise understanding of the role of proinflammatory signaling mechanisms and endogenous regulators remains unknown.
Several studies show that the CBM signalosome (CARMA1 [caspase recruitment domain-containing membrane-associated guanylate kinase protein 1], BCL-10 [B-cell lymphoma 10], and MALT1 [mucosa-associated lymphoid tissue lymphoma translocation protein 1]) mediates NF-κB activation in T and B lymphocytes in response to antigen presentation.25 MALT1 is the key downstream signaling mediator of the CBM signalosome that promotes NF-κB activation via its scaffolding and proteolytic activities.26 Although CARMA1 expression is limited to lymphocytes, an analog, CARMA3, is expressed in endothelial cells.27 Several proinflammatory mediators that signal through G protein-coupled receptors (GPCRs), such as thrombin, angiotensin II, and CXCL8, were shown to stimulate NF-κB activation through the CBM signalosome containing CARMA3.28-30 The role of the CBM signalosome in inflammation beyond the GPCRs is unknown. Gab2 (Grb2 [growth factor receptor-bound protein 2]-associated binding protein 2) is a crucial docking protein that integrates signal transduction evoked by growth factors, interferons, antigen receptors, and cell adhesion molecules.31-34 Our recent studies revealed that Gab2 is a master regulator of multiple inflammatory receptors signaling, including IL-1R1.35 These studies showed that Gab2 is essential to the activation of NF-κB and the prothrombotic gene expression, such as TF, MCP1, and VCAM1 by IL-1β and other proinflammatory mediators.35 Gab2-deficiency was found to attenuate endotoxin- or Streptococcuspneumoniae-induced leukocyte trafficking, generation of thrombin, NETs, and lung injury.35 It is unknown at present whether Gab2 plays a role in the formation of the CBM signalosome to activate NF-κB and induce prothrombotic gene expression. Currently, the potential role of Gab2 and MALT1 in the thrombotic milieu remains unknown.
Here, we show that Gab2 mediates IL-1β–induced mobilization of P-selectin and VWF and expression of TF and VCAM1 through activation of the CBM complex in endothelial cells. Inhibition of MALT1 by silencing the MALT1 gene or with a selective pharmacological inhibitor blocks the IL-1β–induced expression of TF, VCAM1, and exocytosis of VWF and P-selectin. The present studies reveal that the Gab2–MALT1 axis drives the RhoA-mediated exocytosis of VWF and P-selectin, independent of NF-κB activation. In vivo administration of MALT1 inhibitor was shown to confer protection against venous thrombosis in the inferior vena cava (IVC) ligation-induced murine models. Our findings identify a previously unrecognized role of the Gab2–MALT1 axis in the initiation and propagation of coagulation and thromboinflammation and provide novel insights into developing new therapies against venous thromboembolism.
Materials and methods
See supplemental Methods for additional details (available on the Blood Web site).
Cells
Primary human umbilical vein endothelial cells (HUVECs) were obtained from Lonza (Basel, Switzerland) and cultured in the EBM-2 basal medium supplemented with 2% fetal bovine serum and growth supplements at 37°C and 5% CO2 in a humidified incubator. HUVEC passages between 4 and 7 were used in the present study.
Isolation of human neutrophils
Neutrophils from the human blood were isolated based on the protocol described previously.36
Gene silencing, cell treatments, immunoprecipitation, and immunoblotting
HUVECs were transfected with scrambled RNA (scRNA), small interfering RNA (siRNA) specific for Gab2, CARMA3, BCL-10, or MALT1 (200 nM) using Lipofectamine RNAiMAX transfection reagent (Invitrogen). After culturing cells for 48 hours and washing them with serum-free medium, they were stimulated with IL-1β, TNFα, or TNFα/IL-1β as indicated in the Results section and figure legends. The cells were lysed in the sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. An equal amount of protein or volume was subjected to SDS-PAGE, followed by immunoblot analysis to probe for specific signaling proteins or prothrombotic mediators.
For immunoprecipitation studies, the cells were lysed in ice-cold cell lysis buffer (50 mM HEPES, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100) containing protease and phosphatase inhibitors single-use cocktail (Thermo Fisher). Cell lysates were incubated with BCL-10 or CARMA3 antibodies, as specified in the figure legends, overnight at 4°C. The immunocomplexes were pulled down by adding protein A/G agarose beads to the cell lysates incubated with specific antibodies.
Rho activation assay
Rho activation was determined using the Rho activation assay kit (Cytoskeleton Inc., Denver, CO) according to the manufacturer’s instructions by pulldown assay using the Rhotekin–Rho-binding domain (GST-RBD).37
Cell surface enzyme-linked immunosorbent assay (ELISA) and immunofluorescence microscopy
HUVECs cultured in a 96-well plate were used for cell surface ELISA. After experimental treatments, cells were fixed with 4% paraformaldehyde and blocked with 1% bovine serum albumin (BSA). The fixed cells were incubated with P-selectin (5 μg/mL) monoclonal antibody or control IgG for 3 hours at room temperature. After removing the unbound antibody, the cells were washed 3 times with phosphate-buffered saline containing 0.1% BSA and then incubated with horseradish peroxidase-conjugated goat anti-mouse IgG, followed by the substrate tetramethylbenzidine for 30 minutes. The amount of P-selectin antibody bound to the cell surface was determined spectrophotometrically by measuring the absorbance at 650 nm.
The translocation of P-selectin to the cell surface was analyzed by immunofluorescence microscopy using fixed and unpermeabilized cells.
DVT in mice
C57BL/6J male and female mice (10-12 weeks old) were used in these experiments. A well-established IVC ligation-induced flow restriction model (stenosis) of DVT38,39 was used. When mice were administered with the MALT1-specific inhibitor, mepazine (25 mg/kg, pecazine; MedChemExpress), it was given twice intraperitoneally, 2 hours before and 24 hours after the IVC ligation.
Immunohistochemistry
Thin tissue sections of thrombi were incubated with control IgG or antibodies against F4/80 or Ly-6G (5 µg/mL) overnight at 4°C. The sections were then incubated with biotin-labeled goat anti-rat IgG (1:500) and ultrasensitive streptavidin-horse radish peroxidase (1:500) (Sigma; St. Louis, MO) and developed using an 3-amino-9-ethylcarbazole–hydrogen peroxide substrate solution. The sections were counterstained with hematoxylin, mounted, and visualized with an Olympus BX41 microscope.
Data analysis
All experiments were repeated ≥3 times independently. The statistical significance was determined using either one-way ANOVA or Student t test, as appropriate. GraphPad Prism version 8.4.1 software was used for data analysis.
Results
Gab2 regulates IL-1β–induced mobilization of P-selectin and VWF and adhesion of neutrophils to endothelial cells
Endothelial cells contribute to thrombus development through externalization of P-selectin, exocytosis of VWF, and supporting the adhesion of neutrophils.40 Recent studies have shown that IL-1β generated in inflammation promotes venous thrombosis by increasing TF expression, fibrin deposition, leukocyte engagement, NET formation, and pyroptosis.16,41 Consistent with the above studies, we found that IL-1β treatment significantly increased the cell surface expression of P-selectin on endothelial cells (Figure 1A). The inclusion of TNFα with IL-1β further elevated the translocation of P-selectin to the cell surface, much more than IL-1β or TNFα alone (Figure 1A). Therefore, we included TNFα with IL-1β in stimulating endothelial cells when investigating the exocytosis of P-selectin. Since our recent studies35 indicate that Gab2 plays a crucial role in mediating IL-1β–induced inflammatory signaling and the expression of prothrombotic molecules, such as TF and VCAM1 in endothelial cells, we assessed the role of Gab2 in IL-1β–induced exocytosis of P-selectin and VWF. Inhibition of Gab2 expression by siRNA silencing (Figure 1B) markedly suppressed the translocation of P-selectin to the cell surface as measured in a cell surface ELISA (Figure 1C). Gab2 silencing had no effect on total P-selectin protein concentrations (Figure 1B). Corroborating with the above data, immunofluorescence microscopy studies showed immunostaining of P-selectin on the cell surface of intact nonpermeabilized endothelial cells treated with IL-1β but not on cells treated with a control vehicle. Gab2 silencing markedly diminished the cell surface immunostaining of P-selectin in IL-1β–treated endothelial cells (Figure 1D). Parallel studies performed with permeabilized endothelial cells showed no noticeable differences in P-selectin immunostaining among control- and IL-1β–stimulated endothelial cells, either in endothelial cells transfected with scRNA or Gab2 siRNA (Figure 1E). Additional studies showed that IL-1β treatment significantly elevated the secretion of VWF in the endothelial cells, and Gab2 gene silencing blocks the IL-1β–induced secretion of VWF (Figure 1F).
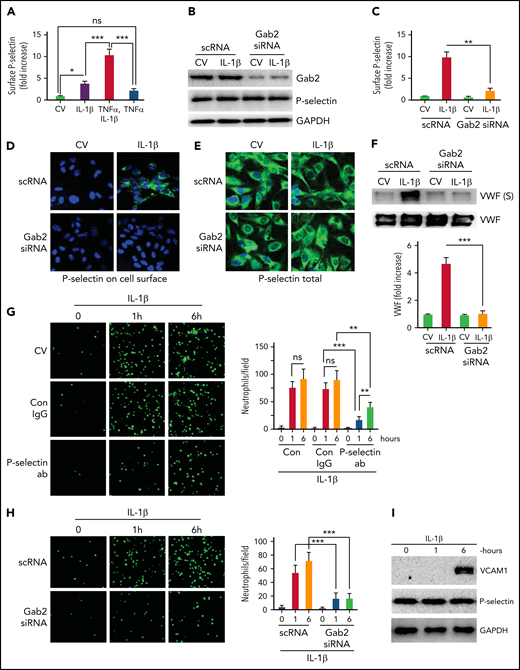
Gab2 is essential for IL-1β–induced exocytosis of P-selectin and VWF in endothelial cells and the adhesion of neutrophils to activated endothelial cells. (A) IL-1β induces P-selectin expression on the endothelial cell surface. HUVECs were treated with IL-1β (10 ng/mL) or TNFα (10 ng/mL) alone or in combination for 1 hour. At the end of 1 hour, the cells were fixed in 2% paraformaldehyde (PFA), and the surface expression of P-selectin was measured by cell surface ELISA. Since the inclusion of TNFα in IL-1β treatment gave a robust increase in P-selectin translocation to the cell surface, in all experiments described in this and other figures that examined the exocytosis of P-selectin and VWF, IL-1β was supplemented with TNFα in treating endothelial cells. (B) Gab2 silencing does not affect total P-selectin concentrations. HUVECs were transfected with 200 nM scrambled RNA (scRNA) or Gab2 small interfering RNA(siRNA). After 48 hours, the cells were treated with IL-1β for 1 hour. The cells were lysed, and the concentrations of Gab2 and P-selectin were analyzed by immunoblotting. Densitometric analysis of immunoblots showed that Gab2 silencing knocked down around 85% of Gab2 and no change in the total P-selectin. (C) Gab2 silencing impairs the translocation of P-selectin to the cell surface. HUVECs transfected with scRNA or Gab2 siRNA were treated with IL-1β for 1 hour. The cell surface expression of P-selectin was measured by cell surface ELISA. (D-E) Gab2 silencing impairs IL-1β–induced expression of P-selectin at the cell surface without impairing the total cellular expression of P-selectin. HUVECs, cultured on cover glasses, were transfected with 200 nM of scRNA or Gab2 siRNA. After 48 hours, the cells were treated with IL-1β for 1 hour. P-selectin expression was analyzed in (D) unpermeabilized or (E) permeabilized cells by immunofluorescence microscopy. (F) Gab2 silencing suppresses IL-1β–induced secretion of VWF. HUVECs transfected with scRNA or Gab2 siRNA were treated with IL-1β for 1 hour. At the end of 1 hour, cell supernatants were removed and precipitated with trichloroacetic acid (TCA) (6%) to concentrate proteins. The suspension of TCA precipitates was subjected to immunoblot analysis to measure secreted (S) VWF. As a control, VWF in cell lysates was also analyzed by immunoblot analysis. Concentrations of secreted VWF were quantified by densitometric analysis of VWF (S) band, and the value obtained in control scRNA transfected cells was arbitrarily assigned as 1. The top panel shows representative immunoblot, and the bottom panel shows densitometric quantification of the VWF (S) band. (G) P-selectin-dependent adhesion of neutrophils to activated endothelial cells. HUVECs cultured on cover glasses were treated with IL-1β for 1 or 6 hours. The cells were washed and incubated with a P-selectin–blocking monoclonal antibody or control IgG (10 µg/mL) for 1 hour. After that, monolayers of HUVECs were layered with PKH-labeled human neutrophils (5 × 105/mL). Neutrophils were allowed to adhere for 30 minutes. Then, unbound neutrophils were removed, the endothelial cell monolayers were washed, and the adhered neutrophils to endothelial cells were fixed in 4% PFA for 15 minutes. The cover glass was mounted with antifade Fluro gel, and the cells were visualized under a confocal microscope at 20× magnification. The micrographs shown were the representative images of 3 independent experiments. The adhered cells were enumerated at 3 random locations in each cover glass, and these data were presented in the right-side panel. (H) Gab2 silencing in endothelial cells attenuates neutrophil adhesion to activated endothelial cells. HUVECs transfected with scRNA or Gab2 siRNA were treated with IL-1β, and the adhesion of neutrophils to activated endothelial cells was analyzed as described in panel (G). (I) IL-1β–induced expression of VCAM1. HUVECs were treated with IL-1β for 1 or 6 hours. The cell lysates were probed for VCAM1 by immunoblot analysis. Data are the mean ± standard deviation of 3 independent experiments. *P < .05, **P < .01, and ***P < .001. ns, no statistically significant difference.
Gab2 is essential for IL-1β–induced exocytosis of P-selectin and VWF in endothelial cells and the adhesion of neutrophils to activated endothelial cells. (A) IL-1β induces P-selectin expression on the endothelial cell surface. HUVECs were treated with IL-1β (10 ng/mL) or TNFα (10 ng/mL) alone or in combination for 1 hour. At the end of 1 hour, the cells were fixed in 2% paraformaldehyde (PFA), and the surface expression of P-selectin was measured by cell surface ELISA. Since the inclusion of TNFα in IL-1β treatment gave a robust increase in P-selectin translocation to the cell surface, in all experiments described in this and other figures that examined the exocytosis of P-selectin and VWF, IL-1β was supplemented with TNFα in treating endothelial cells. (B) Gab2 silencing does not affect total P-selectin concentrations. HUVECs were transfected with 200 nM scrambled RNA (scRNA) or Gab2 small interfering RNA(siRNA). After 48 hours, the cells were treated with IL-1β for 1 hour. The cells were lysed, and the concentrations of Gab2 and P-selectin were analyzed by immunoblotting. Densitometric analysis of immunoblots showed that Gab2 silencing knocked down around 85% of Gab2 and no change in the total P-selectin. (C) Gab2 silencing impairs the translocation of P-selectin to the cell surface. HUVECs transfected with scRNA or Gab2 siRNA were treated with IL-1β for 1 hour. The cell surface expression of P-selectin was measured by cell surface ELISA. (D-E) Gab2 silencing impairs IL-1β–induced expression of P-selectin at the cell surface without impairing the total cellular expression of P-selectin. HUVECs, cultured on cover glasses, were transfected with 200 nM of scRNA or Gab2 siRNA. After 48 hours, the cells were treated with IL-1β for 1 hour. P-selectin expression was analyzed in (D) unpermeabilized or (E) permeabilized cells by immunofluorescence microscopy. (F) Gab2 silencing suppresses IL-1β–induced secretion of VWF. HUVECs transfected with scRNA or Gab2 siRNA were treated with IL-1β for 1 hour. At the end of 1 hour, cell supernatants were removed and precipitated with trichloroacetic acid (TCA) (6%) to concentrate proteins. The suspension of TCA precipitates was subjected to immunoblot analysis to measure secreted (S) VWF. As a control, VWF in cell lysates was also analyzed by immunoblot analysis. Concentrations of secreted VWF were quantified by densitometric analysis of VWF (S) band, and the value obtained in control scRNA transfected cells was arbitrarily assigned as 1. The top panel shows representative immunoblot, and the bottom panel shows densitometric quantification of the VWF (S) band. (G) P-selectin-dependent adhesion of neutrophils to activated endothelial cells. HUVECs cultured on cover glasses were treated with IL-1β for 1 or 6 hours. The cells were washed and incubated with a P-selectin–blocking monoclonal antibody or control IgG (10 µg/mL) for 1 hour. After that, monolayers of HUVECs were layered with PKH-labeled human neutrophils (5 × 105/mL). Neutrophils were allowed to adhere for 30 minutes. Then, unbound neutrophils were removed, the endothelial cell monolayers were washed, and the adhered neutrophils to endothelial cells were fixed in 4% PFA for 15 minutes. The cover glass was mounted with antifade Fluro gel, and the cells were visualized under a confocal microscope at 20× magnification. The micrographs shown were the representative images of 3 independent experiments. The adhered cells were enumerated at 3 random locations in each cover glass, and these data were presented in the right-side panel. (H) Gab2 silencing in endothelial cells attenuates neutrophil adhesion to activated endothelial cells. HUVECs transfected with scRNA or Gab2 siRNA were treated with IL-1β, and the adhesion of neutrophils to activated endothelial cells was analyzed as described in panel (G). (I) IL-1β–induced expression of VCAM1. HUVECs were treated with IL-1β for 1 or 6 hours. The cell lysates were probed for VCAM1 by immunoblot analysis. Data are the mean ± standard deviation of 3 independent experiments. *P < .05, **P < .01, and ***P < .001. ns, no statistically significant difference.
To assess the functional significance of P-selectin expression on the surface, we performed the neutrophil adhesion assay. IL-1β treatment of endothelial cells markedly increased the adhesion of neutrophils to the endothelial monolayer (Figure 1G). No significant differences were observed in neutrophil adhesion to endothelial cells stimulated with IL-1β for either 1 or 6 hours. Pretreatment of IL-1β–stimulated endothelial cells with P-selectin antibodies significantly reduced the adhesion of neutrophils to endothelial cells (Figure 1G). P-selectin antibodies were more effective in reducing neutrophil adhesion to endothelial cells activated for 1 hour. Gab2 silencing, which reduced P-selectin externalization (Figure 1C), suppressed the adhesion of neutrophils to the IL-1β–stimulated endothelial cells (Figure 1H). In additional studies, immunoblot analysis of cell lysates of endothelial cells treated with IL-1β for 1 or 6 hours showed that 1 hour of stimulation was insufficient to induce VCAM1 expression, whereas 6 hours of treatment markedly induced the expression of VCAM1 (Figure 1I). There were no differences in total P-selectin concentrations between unperturbed and IL-1β–stimulated endothelial cells (Figure 1I). Overall, the above data suggest that IL-1β–induced Gab2-mediated surface expression of P-selectin, not VCAM1, plays a role in recruiting neutrophils to the endothelium at the early phase of thromboinflammation.
Gab2-mediated mobilization of P-selectin and VWF are Rho-dependent, and TAK1- and NF-κB–independent
Proinflammatory mediators such as thrombin and histamine mobilize P-selectin and VWF through activation of Rho-dependent pathways.42,43 IL-1β mediates inflammation through Rho-, TAK1-, and NF-κB–dependent pathways.44,45 Our recent studies suggest that Gab2 mediates IL-1β–induced proinflammatory signaling through TAK1- and NF-κB–dependent pathways.35 To investigate the role of Rho-mediated signaling in the IL-1β–induced and Gab2-dependent mobilization of P-selectin and VWF, we first assessed whether IL-1β stimulation induces Rho activation in endothelial cells. As shown in Figure 2A, IL-1β treatment significantly increased the activation of RhoA (increase in Rho-GTP concentrations), and the Rho-GTPase inhibitor, rhosin, completely blocked the IL-1β–induced activation of RhoA. Silencing of Gab2 fully attenuated the IL-1β–induced activation of RhoA (Figure 2B), indicating that IL-1β–induced activation of RhoA is mediated via Gab2. Furthermore, treatment of endothelial cells with Rho-specific inhibitors, rhosin or Y27632, before activating cells with IL-1β, significantly attenuated the IL-1β–induced translocation of P-selectin to the cell surface and VWF secretion into the supernatant medium (Figure 2C). The inhibitors had no effect on total P-selectin or total VWF protein concentrations (Figure 2C).
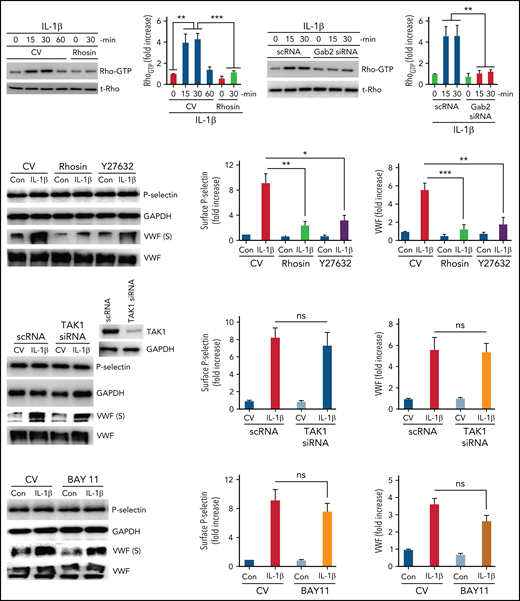
IL-1β–induced and Gab2-dependent mobilization of P-selectin and VWF was Rho-dependent and independent of TAK1 and NF-κB. (A) IL-1β activates Rho kinase in endothelial cells. HUVECs were treated with a control vehicle or Rho-specific inhibitor, rhosin, for 1 hour. After that, the cells were stimulated with IL-1β for indicated periods. Rho activation was measured as GTP-bound Rho using the Rho activation assay kit (left panel). Rho-GTP signals were quantified by densitometric analysis, and the signal intensity in cells not treated with IL-1β or rhosin was taken as 1. (B) Gab2 silencing blocks IL-1β–induced Rho activation in endothelial cells. HUVECs were transfected with 200 nM of scRNA or Gab2 siRNA. After 48 hours, the transfected cells were treated with IL-1β for indicated time periods. Rho activation was measured and quantified as described in (A). (C) Pharmacological inhibition of Rho activation attenuates IL-1β–induced translocation of P-selectin to the cell surface and VWF secretion. HUVECs were treated with a control vehicle or Rho-specific inhibitors, rhosin or Y27632 (10 µM), for 1 hour. Then, the cells were stimulated with IL-1β. After 1 hour, the cell supernatants were collected, and cell lysates were harvested. Cell lysates were probed for total P-selectin, VWF, and GAPDH. Cell supernatants were precipitated with TCA to concentrate proteins and probed for VWF by immunoblot analysis to assess secreted (S) VWF (left panel). The translocation of P-selectin to the cell surface was measured by cell surface ELISA (middle panel). VWF secretion was quantified by densitometric analysis of VWF (S) immunoblots (right panel). (D) TAK1 silencing does not affect IL-1β–induced translocation of P-selectin to the cell surface or VWF secretion. HUVECs were transfected with scRNA or TAK1 siRNA (200 nM) for 48 hours. The transfected cells were stimulated with IL-1β for 1 hour. P-selectin translocation to the cell surface and VWF secretion were evaluated as described in (C). (E) Pharmacological inhibition of NF-κB does not curtail IL-1β–induced translocation of P-selectin to the cell surface or VWF secretion. HUVECs were treated with an NF-κB–specific inhibitor, BAY117082 (20 µM), or a control vehicle for 1 hour. Thereafter, the cells were stimulated with IL-1β for 1 hour, and P-selectin translocation to the cell surface and VWF secretion were evaluated as described in (C). *P < .05, **P < .01, and ***P < .001. ns, no statistically significant difference.
IL-1β–induced and Gab2-dependent mobilization of P-selectin and VWF was Rho-dependent and independent of TAK1 and NF-κB. (A) IL-1β activates Rho kinase in endothelial cells. HUVECs were treated with a control vehicle or Rho-specific inhibitor, rhosin, for 1 hour. After that, the cells were stimulated with IL-1β for indicated periods. Rho activation was measured as GTP-bound Rho using the Rho activation assay kit (left panel). Rho-GTP signals were quantified by densitometric analysis, and the signal intensity in cells not treated with IL-1β or rhosin was taken as 1. (B) Gab2 silencing blocks IL-1β–induced Rho activation in endothelial cells. HUVECs were transfected with 200 nM of scRNA or Gab2 siRNA. After 48 hours, the transfected cells were treated with IL-1β for indicated time periods. Rho activation was measured and quantified as described in (A). (C) Pharmacological inhibition of Rho activation attenuates IL-1β–induced translocation of P-selectin to the cell surface and VWF secretion. HUVECs were treated with a control vehicle or Rho-specific inhibitors, rhosin or Y27632 (10 µM), for 1 hour. Then, the cells were stimulated with IL-1β. After 1 hour, the cell supernatants were collected, and cell lysates were harvested. Cell lysates were probed for total P-selectin, VWF, and GAPDH. Cell supernatants were precipitated with TCA to concentrate proteins and probed for VWF by immunoblot analysis to assess secreted (S) VWF (left panel). The translocation of P-selectin to the cell surface was measured by cell surface ELISA (middle panel). VWF secretion was quantified by densitometric analysis of VWF (S) immunoblots (right panel). (D) TAK1 silencing does not affect IL-1β–induced translocation of P-selectin to the cell surface or VWF secretion. HUVECs were transfected with scRNA or TAK1 siRNA (200 nM) for 48 hours. The transfected cells were stimulated with IL-1β for 1 hour. P-selectin translocation to the cell surface and VWF secretion were evaluated as described in (C). (E) Pharmacological inhibition of NF-κB does not curtail IL-1β–induced translocation of P-selectin to the cell surface or VWF secretion. HUVECs were treated with an NF-κB–specific inhibitor, BAY117082 (20 µM), or a control vehicle for 1 hour. Thereafter, the cells were stimulated with IL-1β for 1 hour, and P-selectin translocation to the cell surface and VWF secretion were evaluated as described in (C). *P < .05, **P < .01, and ***P < .001. ns, no statistically significant difference.
Next, to investigate the role of TAK1 and NF-κB activation in IL-1β–induced Gab2-mediated exocytosis of P-selectin and VWF, endothelial cells were transfected with TAK1 siRNA or treated with a specific NF-κB inhibitor, BAY11-7082, before stimulating with IL-1β. TAK1 siRNA markedly suppressed the expression of TAK1 (Figure 2D). TAK1 silencing had no significant effect on IL-1β–induced P-selectin mobilization or VWF secretion (Figure 2D). Similar results were obtained in cells treated with TAK1-specific inhibitor (5Z)-7-oxozeaenol (data not shown). Similarly, inhibition of NF-κB with BAY11 did not affect the IL-1β–induced externalization of P-selectin or VWF secretion (Figure 2E). The above results suggest that the Gab2/Rho axis and not the Gab2–TAK1–NF-κB axis plays a role in the IL-1β–induced mobilization of P-selectin and VWF in endothelial cells.
MALT1 regulates Rho activation, secretion of prothrombotic mediators, and adhesion of neutrophils
MALT1, a key constituent of the CBM signalosome that plays a crucial role in NF-κB activation, was also shown to regulate Rho activation independently of NF-κB.46 Therefore, we evaluated whether MALT1 is involved in the IL-1β–induced Rho activation and the subsequent exocytosis of P-selectin and VWF in endothelial cells. Inhibition of MALT1 expression by siRNA markedly reduced IL-1β–induced Rho activation in endothelial cells (Figure 3A), suggesting that MALT1 plays a role in the Rho activation. MALT1 silencing also markedly reduced the translocation of P-selectin to the cell surface and secretion of VWF (Figure 3B). MALT1 silencing had no significant effect on total P-selectin and VWF concentrations (Figure 3B). Pharmacological inhibition of MALT1 with a specific inhibitor, mepazine, gave similar results (ie, markedly reduced IL-1β–induced exocytosis of P-selectin and VWF without affecting total P-selectin and VWF concentrations) (Figure 3C). Consistent with the hypothesis that MALT1 plays a crucial role in the IL-1β–induced exocytosis of P-selectin and VWF and the subsequent neutrophil adhesion to activated endothelium, MALT1 silencing or inhibition of MALT1 with a specific pharmacological inhibitor attenuated neutrophil adhesion to IL-1β–activated endothelial cells (Figure 3D-E).
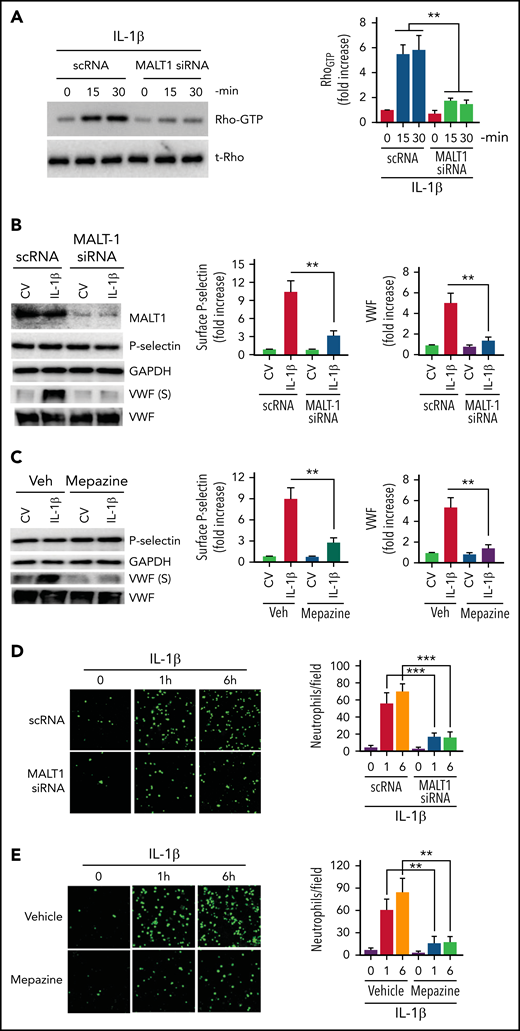
MALT-1 regulates IL-1β–induced Rho activation, mobilization of P-selectin and VWF, and neutrophil adhesion to activated endothelial cells. (A) MALT1 silencing suppresses IL-1β–induced Rho activation in endothelial cells. HUVECs were transfected with 200 nM of scRNA or MALT1 siRNA. After 48 hours, the transfected cells were treated with IL-1β for indicated time periods. Rho activation was measured as GTP-bound Rho as described in the Methods section. The left panel shows a representative blot (t-Rho in the blot indicates total Rho). The right panel shows the quantification of Rho-GTP concentrations from densitometric analysis of signals of immunoblots. Rho-GTP concentrations measured in scRNA-transfected cells and not treated with IL-1β were taken as 1, and other values were shown relative to this value. (B) MALT1 silencing inhibits IL-1β–induced translocation of P-selectin to the cell surface and secretion of VWF. HUVECs transfected with scRNA or MALT1 siRNA were treated with a control vehicle or IL-1β for 1 hour. After 1 hour, the cell supernatants were collected, and cell lysates were harvested. Cell supernatants were precipitated with TCA to concentrate proteins. Cell lysates were probed for MALT1, P-selectin, and VWF by immunoblot analysis; cell supernatants were probed for VWF by immunoblot analysis to assess the concentrations of secreted (S) VWF (left panel). The translocation of P-selectin to the cell surface was measured by cell surface ELISA (middle panel). VWF secretion was quantified by densitometric analysis of VWF (S) immunoblots (right panel). Concentrations of P-selectin on the cell surface and secreted VWF measured in cells transfected with scRNA and treated with a control vehicle were taken as 1.0, and other values were shown relative to these. (C) Pharmacological inhibition of MALT1 attenuates IL-1β–induced P-selectin translocation to the cell surface and secretion of VWF. HUVECs were treated with MALT1 inhibitor, mepazine (20 µM), or vehicle (veh) for 1 hour. After that, the cells were treated with a control vehicle (CV) or IL-1β for 1 hour. The assessment of P-selectin translocation to the cell surface and VWF secretion was performed as described in (B). (D-E) (D) MALT1 silencing or (E) pharmacological inhibition attenuates neutrophil adhesion to IL-1β–activated endothelial cells. HUVECs cultured on cover glasses were transfected with scRNA, MALT1 siRNA, or treated with MALT1 inhibitor, mepazine, as described in the figure legends of (B-C). HUVECs were stimulated with IL-1β for 1 or 6 hours. After washing the cells, PKH-labeled human neutrophils (5 × 105/mL) were added to endothelial cells. After 30 minutes, the unbound neutrophils were removed, and endothelial cells were washed. Neutrophils adhered to endothelial cells were fixed in 4% PFA for 15 minutes. The cells were visualized and imaged under 20× magnification using a confocal microscope (left panel). The cell count enumerated at 3 random locations of each cover glass of 3 independent experiments was shown in the bar graph (right panel). **P < .01 and ***P < .001.
MALT-1 regulates IL-1β–induced Rho activation, mobilization of P-selectin and VWF, and neutrophil adhesion to activated endothelial cells. (A) MALT1 silencing suppresses IL-1β–induced Rho activation in endothelial cells. HUVECs were transfected with 200 nM of scRNA or MALT1 siRNA. After 48 hours, the transfected cells were treated with IL-1β for indicated time periods. Rho activation was measured as GTP-bound Rho as described in the Methods section. The left panel shows a representative blot (t-Rho in the blot indicates total Rho). The right panel shows the quantification of Rho-GTP concentrations from densitometric analysis of signals of immunoblots. Rho-GTP concentrations measured in scRNA-transfected cells and not treated with IL-1β were taken as 1, and other values were shown relative to this value. (B) MALT1 silencing inhibits IL-1β–induced translocation of P-selectin to the cell surface and secretion of VWF. HUVECs transfected with scRNA or MALT1 siRNA were treated with a control vehicle or IL-1β for 1 hour. After 1 hour, the cell supernatants were collected, and cell lysates were harvested. Cell supernatants were precipitated with TCA to concentrate proteins. Cell lysates were probed for MALT1, P-selectin, and VWF by immunoblot analysis; cell supernatants were probed for VWF by immunoblot analysis to assess the concentrations of secreted (S) VWF (left panel). The translocation of P-selectin to the cell surface was measured by cell surface ELISA (middle panel). VWF secretion was quantified by densitometric analysis of VWF (S) immunoblots (right panel). Concentrations of P-selectin on the cell surface and secreted VWF measured in cells transfected with scRNA and treated with a control vehicle were taken as 1.0, and other values were shown relative to these. (C) Pharmacological inhibition of MALT1 attenuates IL-1β–induced P-selectin translocation to the cell surface and secretion of VWF. HUVECs were treated with MALT1 inhibitor, mepazine (20 µM), or vehicle (veh) for 1 hour. After that, the cells were treated with a control vehicle (CV) or IL-1β for 1 hour. The assessment of P-selectin translocation to the cell surface and VWF secretion was performed as described in (B). (D-E) (D) MALT1 silencing or (E) pharmacological inhibition attenuates neutrophil adhesion to IL-1β–activated endothelial cells. HUVECs cultured on cover glasses were transfected with scRNA, MALT1 siRNA, or treated with MALT1 inhibitor, mepazine, as described in the figure legends of (B-C). HUVECs were stimulated with IL-1β for 1 or 6 hours. After washing the cells, PKH-labeled human neutrophils (5 × 105/mL) were added to endothelial cells. After 30 minutes, the unbound neutrophils were removed, and endothelial cells were washed. Neutrophils adhered to endothelial cells were fixed in 4% PFA for 15 minutes. The cells were visualized and imaged under 20× magnification using a confocal microscope (left panel). The cell count enumerated at 3 random locations of each cover glass of 3 independent experiments was shown in the bar graph (right panel). **P < .01 and ***P < .001.
Since our recent studies showed that IL-1β–induced Gab2-mediated cell signaling also leads to NF-κB activation and the expression of prothrombotic mediators, such as TF and VCAM1,35 we also looked at the role of MALT1 in IL-1β–induced activation of NF-κB and expression of TF and VCAM1. Silencing of MALT1 notably attenuated IL-1β–induced activation of p65 (Figure 4A) and expression of TF and VCAM1 (Figure 4B). Consistent with the above data, treatment of endothelial cells with MALT1 specific inhibitor, mepazine, markedly suppressed the IL-1β–induced activation of NF-κB (Figure 4C) and the expression of TF and VCAM1 (Figure 4D). Similar results were obtained with MI-2, another pharmacological inhibitor of MALT1 (data not shown).
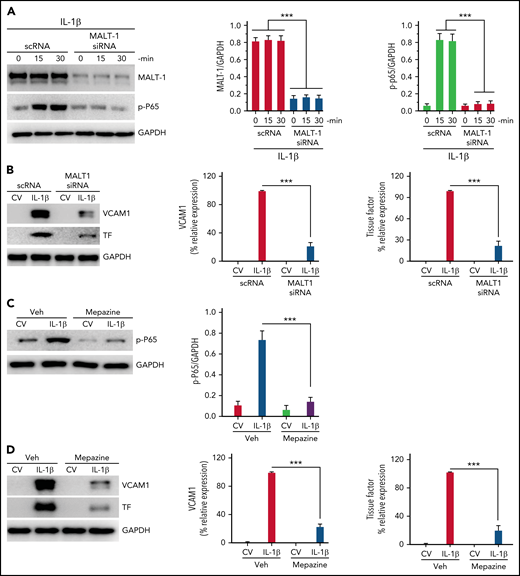
MALT1 inhibition suppresses IL-1β–induced thromboinflammatory gene expression. (A) MALT1 silencing suppresses IL-1β–induced NF-κB activation. HUVECs were transfected with scRNA or MALT1 siRNA (200 nM). After 48 hours, the transfected cells were stimulated with IL-1β for varying times. At the end of treatment, cell lysates were harvested, and the phosphorylation of the P65 subunit of NF-κB was analyzed by immunoblot analysis. The blot was also probed for MALT1 to confirm MALT1 knockdown and GAPDH as a loading control. The left panel shows a representative immunoblot, and the middle and right panels show quantification of MALT1 knockdown and p65 phosphorylation by densitometric analysis of immunoblots. (B) MALT1 silencing suppresses IL-1β–induced expression of TF and VCAM1. HUVECs transfected with scRNA or MALT1 siRNA were treated with a control vehicle (CV) or IL-1β for 6 hours. The cell lysates were probed for VCAM1 and TF expression by immunoblot analysis. The left panel shows a representative immunoblot, and the middle and right panels show quantification of VCAM1 and TF concentrations by densitometric analysis of immunoblots. The concentrations of VCAM1 and TF measured in IL-1β–stimulated cells were taken as 100%, and other values were shown relative to these values. (C-D) (C) MALT1 inhibitor attenuates IL-1β–induced NF-κB activation and (D) expression of VCAM1 and TF. HUVEC were treated with MALT1-specific inhibitor mepazine (20 µM) for 1 hourand then the cells were treated with a control vehcile (CV) or IL-1β for 30 minutes (C) or 6 hours (D). The cell lysates were probed for the phosphorylation of p65 (C) or the expression of VCAM1 and TF (D). The left panel shows a representative immunoblot, and the middle and right panels show densitometric quantification of immunoblot band intensities. The concentrations of p65 were normalized to GAPDH (C). VCAM1 and TF concentrations measured in IL-1β–stimulated cells were taken as 100%, and other values were shown relative to these values. Data are the mean ± standard deviation of 3 independent experiments. ***P < .001.
MALT1 inhibition suppresses IL-1β–induced thromboinflammatory gene expression. (A) MALT1 silencing suppresses IL-1β–induced NF-κB activation. HUVECs were transfected with scRNA or MALT1 siRNA (200 nM). After 48 hours, the transfected cells were stimulated with IL-1β for varying times. At the end of treatment, cell lysates were harvested, and the phosphorylation of the P65 subunit of NF-κB was analyzed by immunoblot analysis. The blot was also probed for MALT1 to confirm MALT1 knockdown and GAPDH as a loading control. The left panel shows a representative immunoblot, and the middle and right panels show quantification of MALT1 knockdown and p65 phosphorylation by densitometric analysis of immunoblots. (B) MALT1 silencing suppresses IL-1β–induced expression of TF and VCAM1. HUVECs transfected with scRNA or MALT1 siRNA were treated with a control vehicle (CV) or IL-1β for 6 hours. The cell lysates were probed for VCAM1 and TF expression by immunoblot analysis. The left panel shows a representative immunoblot, and the middle and right panels show quantification of VCAM1 and TF concentrations by densitometric analysis of immunoblots. The concentrations of VCAM1 and TF measured in IL-1β–stimulated cells were taken as 100%, and other values were shown relative to these values. (C-D) (C) MALT1 inhibitor attenuates IL-1β–induced NF-κB activation and (D) expression of VCAM1 and TF. HUVEC were treated with MALT1-specific inhibitor mepazine (20 µM) for 1 hourand then the cells were treated with a control vehcile (CV) or IL-1β for 30 minutes (C) or 6 hours (D). The cell lysates were probed for the phosphorylation of p65 (C) or the expression of VCAM1 and TF (D). The left panel shows a representative immunoblot, and the middle and right panels show densitometric quantification of immunoblot band intensities. The concentrations of p65 were normalized to GAPDH (C). VCAM1 and TF concentrations measured in IL-1β–stimulated cells were taken as 100%, and other values were shown relative to these values. Data are the mean ± standard deviation of 3 independent experiments. ***P < .001.
IL-1β induces the activation of CARMA3 via Gab2/PLCγ2/PKC axis
The engagement of GPCRs and lymphocyte receptors triggers the activation of PKCβ/θ and the phosphorylation of CARMA3 and initiates the formation of the CBM signaling complex.27 Several growth factors and ILs activate PLCγ/PKC signaling via Gab2.47,48 Therefore, we assessed whether IL-1β induces the phosphorylation of CARMA3 in endothelial cells and the role of Gab2 and PLCγ/PKC signaling in IL-1β–induced activation of CARMA3. As shown in Figure 5A, IL-1β treatment induced the phosphorylation of CARMA3. The knockdown of Gab2 or PLCγ2 expression by siRNA silencing reduced the IL-1β–induced phosphorylation of CARMA3 (Figure 5B). Additional studies showed that IL-1β treatment markedly induced the phosphorylation of PLCγ2 and PKC isoforms (PKCα/β and PKCδ) (Figure 5C). Gab2 silencing attenuated the IL-1β–induced phosphorylation of PLCγ2, PKCα/β, and PKCδ (Figure 5D). Additional experiments revealed that PLCγ2 silencing completely inhibited the IL-1β–induced activation of PKCα/β and PKCδ (Figure 5E), indicating that IL-1β–induced PLCγ2 activation is upstream of PKC activation and downstream of Gab2. Treatment of endothelial cells with PKC-specific inhibitors, such as Go6976 and BisindoM-1, markedly reduced IL-1β–induced phosphorylation of CARMA3 (Figure 5F). Overall, the above data provide compelling evidence that the Gab2/PLCγ2/PKC axis mediates IL-1β–induced activation of CARMA3.
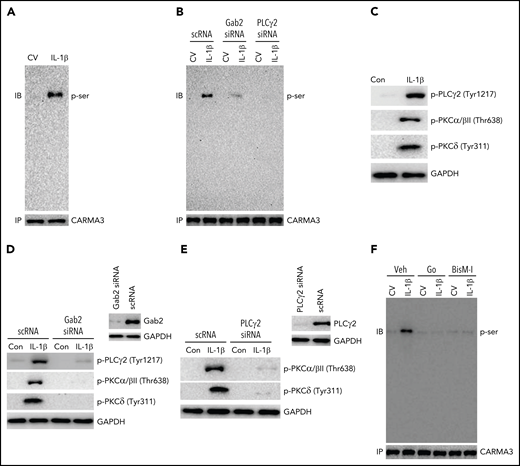
Gab2/PLCγ2/PKC axis mediates IL-1β–induced activation of CARMA3. (A) IL-1β treatment induces the phosphorylation of CARMA3 in endothelial cells. HUVEC were treated with a control vehicle or IL-1β for 5 minutes. The cells were lysed in radioimmunoprecipitation assay (RIPA) buffer, and the CARMA3 was immunoprecipitated using polyclonal CARMA3 antibodies. The immunoprecipitates were subjected to immunoblot analysis and probed with monoclonal antibodies against phosphoserine. (B) Gab2 or PLCγ2 silencing blocks IL-1β–induced phosphorylation of CARMA3. HUVECs transfected with scRNA, Gab2 siRNA, or PLCγ2 siRNA were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were immunoprecipitated with CARMA3 antibodies, and the immunoprecipitates were probed for the presence of phosphorylated CARMA3, as described in the legend to panel (A). (C) IL-1β induces the activation of PLCγ2 and PKC isomers in endothelial cells. HUVECs were treated with a control vehicle or IL-1β for 5 minutes. At the end of the 5-minute treatment, the cell lysates were harvested and probed for the phosphorylation of PLCγ2, PKCα/β, and PKCδ by immunoblot analysis using antibodies against phosphorylated PLCγ2, PKCα/β, and PKCδ. (D) Gab2 silencing inhibits IL-1β–induced activation of PLCγ2 and PKC isomers in endothelial cells. HUVECs were transfected with scRNA or Gab2 siRNA (200 nM). After 48 hours, the Gab2 knockdown was confirmed by immunoblot analysis. HUVECs transfected with scRNA or Gab2 siRNA were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were probed for the presence of phosphorylated PLCγ2, PKCα/β, and PKCδ by immunoblot analysis. (E) PLCγ2 silencing blocks the IL-1β–induced activation of PKC isomers. HUVECs were transfected with scRNA or PLCγ2 siRNA. After 48 hours, PLCγ2 knockdown was confirmed by immunoblotting. HUVECs transfected with scRNA or PLCγ2 siRNA were treated with a control vehicle or IL-1β for 5 minutes. The phosphorylation of PKCα/β and PKCδ were analyzed by immunoblot analysis. (F) Inhibition of PKC attenuates IL-1β–induced phosphorylation of CARMA3. HUVECs were treated with PKC α/β inhibitor Go6976 (Go; 100 nM), pan-PKC inhibitor bisindolylmaleimide 1 (BisM-1; 500 nM), or DMSO vehicle for 1 hour. After that, the cells were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were processed as described in the panel (D) and probed for the phosphorylation of CARMA3. The immunoblots shown were the representative blots of 3 independent experiments with similar results.
Gab2/PLCγ2/PKC axis mediates IL-1β–induced activation of CARMA3. (A) IL-1β treatment induces the phosphorylation of CARMA3 in endothelial cells. HUVEC were treated with a control vehicle or IL-1β for 5 minutes. The cells were lysed in radioimmunoprecipitation assay (RIPA) buffer, and the CARMA3 was immunoprecipitated using polyclonal CARMA3 antibodies. The immunoprecipitates were subjected to immunoblot analysis and probed with monoclonal antibodies against phosphoserine. (B) Gab2 or PLCγ2 silencing blocks IL-1β–induced phosphorylation of CARMA3. HUVECs transfected with scRNA, Gab2 siRNA, or PLCγ2 siRNA were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were immunoprecipitated with CARMA3 antibodies, and the immunoprecipitates were probed for the presence of phosphorylated CARMA3, as described in the legend to panel (A). (C) IL-1β induces the activation of PLCγ2 and PKC isomers in endothelial cells. HUVECs were treated with a control vehicle or IL-1β for 5 minutes. At the end of the 5-minute treatment, the cell lysates were harvested and probed for the phosphorylation of PLCγ2, PKCα/β, and PKCδ by immunoblot analysis using antibodies against phosphorylated PLCγ2, PKCα/β, and PKCδ. (D) Gab2 silencing inhibits IL-1β–induced activation of PLCγ2 and PKC isomers in endothelial cells. HUVECs were transfected with scRNA or Gab2 siRNA (200 nM). After 48 hours, the Gab2 knockdown was confirmed by immunoblot analysis. HUVECs transfected with scRNA or Gab2 siRNA were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were probed for the presence of phosphorylated PLCγ2, PKCα/β, and PKCδ by immunoblot analysis. (E) PLCγ2 silencing blocks the IL-1β–induced activation of PKC isomers. HUVECs were transfected with scRNA or PLCγ2 siRNA. After 48 hours, PLCγ2 knockdown was confirmed by immunoblotting. HUVECs transfected with scRNA or PLCγ2 siRNA were treated with a control vehicle or IL-1β for 5 minutes. The phosphorylation of PKCα/β and PKCδ were analyzed by immunoblot analysis. (F) Inhibition of PKC attenuates IL-1β–induced phosphorylation of CARMA3. HUVECs were treated with PKC α/β inhibitor Go6976 (Go; 100 nM), pan-PKC inhibitor bisindolylmaleimide 1 (BisM-1; 500 nM), or DMSO vehicle for 1 hour. After that, the cells were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were processed as described in the panel (D) and probed for the phosphorylation of CARMA3. The immunoblots shown were the representative blots of 3 independent experiments with similar results.
Gab2 facilitates the formation of IL-1β–induced CBM signalosome and MALT1 interaction with TRAF6
In the CBM signalosome, MALT1 acts as a scaffold protein to activate IKK (inhibitor of NF-κβ kinase)/NF-κB transcription factors.25,49 MALT1 interacts with TRAF6 and ubiquitinates IKK subunit NF-κβ essential modulator for subsequent activation of NF-κB.50 It is well-established that TRAF6 is a central regulator of IL-1β–induced activation of NF-κB.51,52 Consistent with the hypothesis that IL-1β–induced activation of NF-κB involves the formation of CBM signalosome and the association of MALT1 with TRAF6, we found an increased association of CARMA3 and MALT1 with BCL-10 (Figure 6A) and MALT1 with TRAF6 (Figure 6B) in endothelial cells stimulated with IL-1β. Next, to assess the role of Gab2 in the IL-1β–induced formation of the CBM signalosome and the MALT1-TRAF6 complex formation, we knocked down the Gab2 in the endothelial cells and analyzed the IL-1β–induced formation of the CBM signalosome and association of MALT1 with TRAF6. As shown in Figure 6C, Gab2 silencing blocked the interaction of CARMA3 and MALT1 with BCL-10 (Figure 6C). Gab2 silencing also suppressed the interaction of MALT1 with TRAF6 (Figure 6D). Overall, the above data indicate that Gab2 orchestrates the assembly of CARMA3–BCL-10–MALT1 and the subsequent interaction of MALT1 with TRAF6.
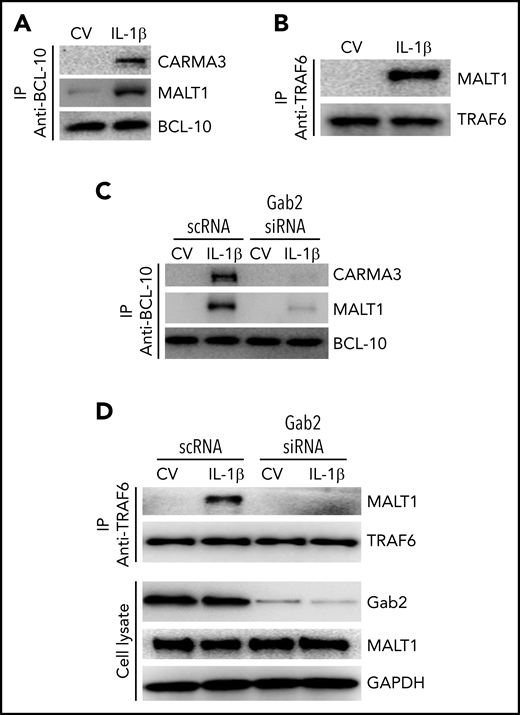
Gab2 facilitates IL-1β–induced assembly of the CBM signalosome and MALT1 interaction with TRAF6. (A) IL-1β induces the formation of the CBM signalosome. HUVECs were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were immunoprecipitated with BCL-10 antibodies, and the immunoprecipitates were probed for the presence of CARMA3, MALT1, and BCL-10 by immunoblot analysis. (B) IL-1β induces MALT1 association with TRAF6. HUVECs were treated with a control vehicle or IL-1β for 5 minutes, and the cell lysates were immunoprecipitated with TRAF6 antibodies. The immunoprecipitates were probed for the presence of MALT1 and TRAF6. (C) Gab2 silencing prevents IL-1β–induced CBM signalosome formation. HUVECs were transfected with scRNA or Gab2 siRNA (200 nM). After 48 hours, the cells were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were immunoprecipitated with BCL-10 antibodies, and the immunoprecipitates were probed for the presence of CARMA3, MALT1, and BCL-10 by immunoblot analysis. (D) Gab2 silencing abolishes IL-1β–induced MALT1 association with TRAF6. HUVECs transfected with scRNA or Gab2 siRNA were treated with a control vehicle of IL-1β for 5 minutes. The cell lysates were immunoprecipitated with TRAF6 antibodies, and the immunoprecipitates were probed for the presence of MALT1 and TRAF6. In addition, cell lysates were probed for Gab2, MALT1, and GAPDH.
Gab2 facilitates IL-1β–induced assembly of the CBM signalosome and MALT1 interaction with TRAF6. (A) IL-1β induces the formation of the CBM signalosome. HUVECs were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were immunoprecipitated with BCL-10 antibodies, and the immunoprecipitates were probed for the presence of CARMA3, MALT1, and BCL-10 by immunoblot analysis. (B) IL-1β induces MALT1 association with TRAF6. HUVECs were treated with a control vehicle or IL-1β for 5 minutes, and the cell lysates were immunoprecipitated with TRAF6 antibodies. The immunoprecipitates were probed for the presence of MALT1 and TRAF6. (C) Gab2 silencing prevents IL-1β–induced CBM signalosome formation. HUVECs were transfected with scRNA or Gab2 siRNA (200 nM). After 48 hours, the cells were treated with a control vehicle or IL-1β for 5 minutes. The cell lysates were immunoprecipitated with BCL-10 antibodies, and the immunoprecipitates were probed for the presence of CARMA3, MALT1, and BCL-10 by immunoblot analysis. (D) Gab2 silencing abolishes IL-1β–induced MALT1 association with TRAF6. HUVECs transfected with scRNA or Gab2 siRNA were treated with a control vehicle of IL-1β for 5 minutes. The cell lysates were immunoprecipitated with TRAF6 antibodies, and the immunoprecipitates were probed for the presence of MALT1 and TRAF6. In addition, cell lysates were probed for Gab2, MALT1, and GAPDH.
MALT1 inhibition attenuates DVT in mice
Our in vitro studies in endothelial cells show that MALT1 plays a crucial role in IL-1β–induced expression of TF and VCAM1, exocytosis of P-selectin and VWF, and neutrophil recruitment. All the above events are known to play crucial roles in the pathogenesis of DVT.10,13 Therefore, we investigated the effect of MALT1 inhibition in thrombus development in DVT. In the first set of experiments, we used the IVC ligation-induced flow-restriction stenosis model to induce thrombosis. Briefly, wild-type mice were intraperitoneally administered with mepazine, a pharmacological inhibitor of MALT1, with a cumulative dose of 25 mg/kg 2 hours before and 24 hours after the IVC ligation-induced flow-restriction stenosis. Analysis of thrombus formation at 48 hours following the IVC ligation showed that 90% of animals treated with a control vehicle developed thrombus of variable lengths and weights (Figure 7A-B). In contrast, only 30% of animals developed thrombus in mepazine-treated animals following the IVC ligation (Figure 7A-B). Thrombus length and weights were significantly lower in mepazine-treated animals compared with vehicle-treated animals (Figure 7C-D). These data clearly illustrate that the MALT1 inhibitor significantly reduces the IVC ligation-induced venous thrombosis. In additional studies, we analyzed the accumulation of neutrophils and monocytes in the thrombus by immunostaining of thrombus with Ly-6G and F4/80 antibodies, respectively. We found extensive accumulation of Ly-6G+ and F4/80+ cells in the thrombus, and mepazine treatment significantly inhibited the accumulation of Ly-6G+ and F4/80+ cells in the thrombus (Figure 7E-F).
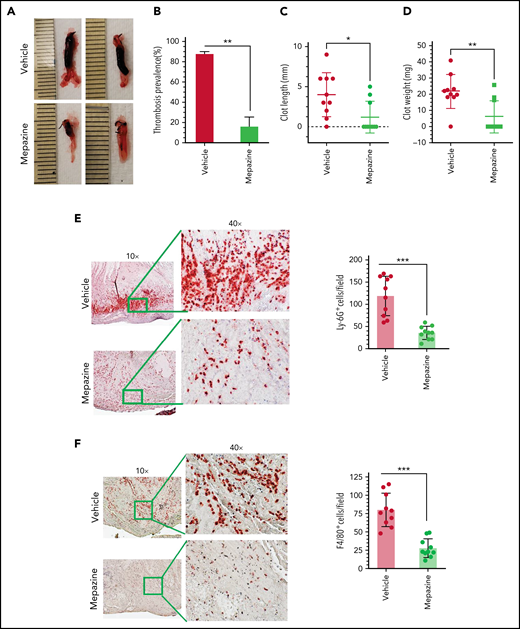
MALT1 inhibition protects against venous thrombosis induced by flow restriction. C57/BL 6J wild-type mice were administered 2 doses of MALT1 inhibitor, mepazine (12.5 mg/kg body weight each dose, intraperitoneally), the first dose 2 hours before surgery, and a second dose 24 hours after the IVC ligation. Control mice were administered with a control vehicle (DMSO). Mice were subjected to the IVC ligation-induced stenosis. Forty-eight hours following the IVC ligation, mice were killed, and thrombus formation in the ligated vein was evaluated. (A) Representative images of thrombus; (B) thrombosis prevalence; (C) thrombus length; (D) thrombus weight. Thrombi collected were processed for tissue sectioning, and sections were stained with antibodies against (E) Ly-6G or (F) F4/80 antigens. The sections were visualized under a bright field microscope at 10×, and selected areas (boxed) were viewed at 40× magnification. The number of LY-6G or F4/80-positive cells was analyzed at 10 randomly chosen areas of the high-power field covering the entire section of 10 different mice. Data are mean ± standard deviation of 3 independent experiments. *P < .05, **P < .01, and ***P < .001.
MALT1 inhibition protects against venous thrombosis induced by flow restriction. C57/BL 6J wild-type mice were administered 2 doses of MALT1 inhibitor, mepazine (12.5 mg/kg body weight each dose, intraperitoneally), the first dose 2 hours before surgery, and a second dose 24 hours after the IVC ligation. Control mice were administered with a control vehicle (DMSO). Mice were subjected to the IVC ligation-induced stenosis. Forty-eight hours following the IVC ligation, mice were killed, and thrombus formation in the ligated vein was evaluated. (A) Representative images of thrombus; (B) thrombosis prevalence; (C) thrombus length; (D) thrombus weight. Thrombi collected were processed for tissue sectioning, and sections were stained with antibodies against (E) Ly-6G or (F) F4/80 antigens. The sections were visualized under a bright field microscope at 10×, and selected areas (boxed) were viewed at 40× magnification. The number of LY-6G or F4/80-positive cells was analyzed at 10 randomly chosen areas of the high-power field covering the entire section of 10 different mice. Data are mean ± standard deviation of 3 independent experiments. *P < .05, **P < .01, and ***P < .001.
In the second set of experiments, we used the IVC ligation-induced stasis model to evaluate the therapeutic efficacy of mepazine in mice. In this model, all mice treated with a control vehicle developed a thrombus following the IVC ligation (supplemental Figure 1 in the data supplement). Administration of a single dose of mepazine (12.5 mg/kg) to mice before the IVC ligation markedly reduced thrombus formation. Thrombus length and weights were decreased by 85% and 90%, respectively, compared with that observed in control vehicle-treated animals (supplemental Figure 1B-C). Administration of mepazine to mice at 4 hours after following the IVC ligation also significantly reduced the stasis-induced thrombosis (supplemental Figure 1B-C). Additional experiments in which various prothrombotic mediators in the thrombi were analyzed showed higher concentrations of IL-1β, TF, VCAM1, CD41 (platelet marker), and fibrin in vehicle-treated mice and a significant decrease of them in mepazine-treated mice (supplemental Figure 2A-B).
MALT1 inhibition attenuates NET formation in vitro and in vivo
MALT1 is expressed by many cell types, including endothelial cells, neutrophils, and monocytes.53 Since neutrophils and associated NETs play a critical role in thrombogenesis,22,23 we analyzed the NET formation in the thrombus by coimmunostaining thrombus sections with citH3 and Ly6G antibodies. We found a notable presence of NETs in thrombi formed in the IVC ligation-induced stenosis of mice administered with a control vehicle. Mepazine treatment markedly suppressed the NET formation in the thrombus (supplemental Figure 3A).
Since mepazine treatment also decreased neutrophil accumulation in the thrombus, it is unclear whether the decreased NET formation was because of a reduction in neutrophil accumulation or the direct effect of mepazine on neutrophils on NET formation. To address this, in vitro experiments were performed where human neutrophils were treated with a control vehicle or mepazine and stimulated with PMA or IL-1β/TNFα to induce NET formation. Both PMA and IL-1β/TNFα treatments significantly induced the NET formation, as evidenced by increased citH3 and colocalization with MPO (supplemental Figure 3B-C). Treatment of neutrophils with mepazine significantly lowered the PMA or IL-1β/TNFα-induced NET formation (supplemental Figure 3B-C). Overall, our results suggest that the reduction of NETs in the thrombus in mepazine-treated mice could have come from both reduction in neutrophil accumulation and inhibition of NETosis.
Gab2 deficiency attenuates venous thrombosis in mice
Since our in vitro data convincingly show that Gab2 regulates the assembly of CARMA3/BCL10/MALT1 complex and MALT1 interaction with downstream signaling molecules, we next examined whether Gab2 deficiency protects against the IVC ligation-induced thrombosis as observed in mice treated with MALT1 pharmacological inhibitor. Thrombus formation was significantly lower in Gab2−/− mice compared with their littermate control subjects (supplemental Figure 4A-C). As observed in mice treated with MALT1 inhibitor, Gab2 deficiency led to a marked reduction in neutrophil and macrophage accumulation in thrombi (supplemental Figure 4D-E).
Discussion
The endothelium is an important contributor to thromboinflammation in DVT.11,16,40 The activation of endothelium precedes the accumulation of platelets and leukocytes in growing thrombus in DVT.11,54 Our current study identifies that the Gab2–CBM axis plays a crucial role in the propagation of IL-1β–induced prothrombotic mediators, such as expression of TF, VCAM1, and exocytosis of P-selectin and VWF by modulating 2 distinctive signaling pathways, NF-κB- and Rho-mediated pathways. Inhibition of MALT1 protease activity in the CBM signalosome by a specific MALT1 pharmacological inhibitor attenuates IL-1β–induced expression of prothrombotic mediators in the endothelial cell model system and attenuates the IVC ligation-induced venous thrombosis in a murine model. Identification of novel regulators of IL-1β–induced prothrombotic vascular cell signaling may be helpful in intervening in thrombus development.
The cell surface expression of VCAM1 and P-selectin on the endothelium confers the adhesion of neutrophils to the endothelium in vascular dysfunction.55,56 Here, we found that treatment of endothelial cells with IL-1β led to a marked increase in cell surface expression of P-selectin and VWF secretion through exocytosis. Consistent with the data that cell surface expression of P-selectin plays a crucial role in the adhesion of neutrophils to activated endothelial cells, functional blockade of P-selectin with an antibody markedly reduced the adhesion of neutrophils to IL-1β–treated endothelial cells. It may be interesting to note that the P-selectin antibody is more effective in preventing neutrophil adhesion to activated endothelial cells at the early stage of activation (in cells treated with IL-1β for 1 hour) compared with a late stage (in cells treated with IL-1β for 6 hours) despite similar concentrations of P-selectin expression on the cell surface at both times. It is likely that IL-1β–induced expression of VCAM1 or other adhesive receptors at the cell surface could substitute P-selectin for neutrophil adhesion to the endothelium at a late stage of endothelial cell activation. Consistent with this possibility, no expression of VCAM1 was found in endothelial cells treated with IL-1β for 1 hour, whereas marked expression of VCAM1 was found at 6 hours of IL-1β treatment. Gab2 silencing, which inhibited both IL-1β–induced externalization of P-selectin to the cell surface and de novo induction of VCAM1, markedly reduced neutrophil adhesion to endothelial cells at both early and late stages of endothelial cell activation.
IL-1β activates RhoA, TAK1, and NF-κB signaling pathways and induces inflammatory gene expression.57,58 Rho activation was shown to play a role in histamine and thrombin-induced exocytosis of P-selectin and VWF.42,43 Gab2 was shown to activate RhoA in cancer cells stimulated with ILs or growth factors.48,59,60 Gab2 activation of RhoA in endothelial cells and its relevance in thromboinflammation is unknown. Our present study showed that Gab2 knockdown blocked IL-1β activation of RhoA in endothelial cells. Furthermore, inhibition of RhoA and not TAK1 or NF-κB markedly attenuated the IL-1β–induced mobilization of P-selectin and VWF secretion. However, inhibition of TAK1 or NF-κB fully blocked the expression of TF and VCAM1 (data not shown). These data suggest that the Gab2–RhoA axis regulates IL-1β–induced translocation of P-selectin and exocytosis of VWF in endothelial cells independent of TAK1 and NF-κB, whereas the Gab2–TAK–NF-κB axis plays an essential role in IL-1β–induced expression of TF and VCAM1.
MALT1 is an enzymatic signaling adapter protein50 and is known to play a crucial role in the GPCR-induced activation of NF-κB in endothelial cells.49,50 MALT1 activation is also known to induce endothelial barrier disruption via the activation of RhoA.46 Our current studies reveal that MALT1-mediated RhoA activation in endothelial cells contributes to the mobilization of P-selectin to the cell surface and exocytosis of VWF as MALT1 silencing or inhibition by a pharmacological inhibitor-blocked IL-1β–induced RhoA activation and attenuated the externalization of P-selectin, VWF secretion, and adhesion of neutrophils to endothelial cells. Consistent with the previous studies,28,29,49 MALT1 silencing or pharmacological inhibition suppressed the activation of NF-κB and expression of TF and VCAM1 in endothelial cells. However, as reported earlier,61,62 we found that IL-1β–induced activation of NF-κB in endothelial was independent of Rho activation. Rho-specific inhibitors had no effect on IL-1β–induced activation of NF-κB or the induction of TF and VCAM1 (data not shown). Overall, these data suggest that IL-1β concomitantly activates divergent Rho and NF-κB signaling to mobilize and induce the expression of prothrombotic mediators, and MALT1 plays a crucial role in propagating both pathways.
MALT1 is a terminal effector of the CBM complex, which interacts with TRAF6 and activates the IKK complex and NF-κB.49,50 The CARMA1 and CARMA3 activate MALT1 in lymphocytes and endothelial cells, respectively.27-29,50 The phosphorylation of CARMA3 is a crucial event that initiates the assembly of the CBM complex and the propagation of downstream signaling.63,64 The PKCs phosphorylate CARMA1/3 and trigger the assembly of the CBM complex.27,65 IL-1β was shown to transduce the inflammatory signaling via PLCγ/PKC axis.66,67 It is previously unknown whether IL-1β activates CARMA3 and the role of the Gab2/PLCγ/PKC axis in this process. In the current study, we found that IL-1β phosphorylates CARMA3, and silencing of either Gab2 or PLCγ2 blocked the phosphorylation of CARMA3. Our studies also showed that IL-1β phosphorylates PLCγ and downstream PKC isomers, PKCα/β/δ, in endothelial cells, and the silencing of Gab2 blocks the IL-1β–induced phosphorylation of PLCγ2/PKCs. Pharmacological inhibition of PKC suppressed IL-1β–induced phosphorylation of CARMA3. The above data suggest that the Gab2/PLCγ2/PKC axis plays a crucial role in the IL-1β–induced activation of CARMA3.
The phosphorylation of CARMA3 initiates the assembly of the CBM signalosome and activation of MALT1.29,50,64 Formation of the CBM signalosome leads to activation of NF-κB in lymphocytes and endothelial cells.68 MALT1 in the CBM signalosome interacts with TRAF6.49,63 Previous studies showed that TRAF6 plays a pivotal role in the IL-1β–induced cell signaling.51,69 However, there was no information on the involvement of the CBM signalosome or MALT1 in IL-1β–induced signaling. The data presented in the current manuscript provide convincing evidence that IL-1β treatment induces the assembly of the CBM complex and interaction of MALT1 with TRAF6. More importantly, Gab2 silencing blocked the assembly of the CBM complex and MALT1–TRAF6 interactions, indicating that Gab2 facilitates the above interactions induced by IL-1β. Overall, our data identify a novel role for the Gab2-PLCγ2 axis in forming the CBM signalosome in endothelial cells and the essential role of MALT1 in mediating various signaling pathways induced by IL-1β.
Earlier studies suggested that the involvement of the CBM complex in NF-κB activation in endothelial cells is specific to GPCR agonists as the knockdown of CARMA3, BCL-10, or MALT1 failed to block TNFα- or IL-1β–induced activation of NF-κB.28 However, contrary to the above data, our current studies provide strong evidence that IL-1β–induced activation of NF-κB also involves the action of the CBM complex as the silencing of either MALT1 or BCL-10 (data not shown) markedly reduced IL-1β–induced activation of NF-κB and NF-κB–mediated expression of prothrombotic mediators TF and VCAM1. Currently, the reasons for the above discrepancy are unknown but may be because of differences in cell model systems or experimental conditions. The earlier study28 employed a mouse endothelial cell line (SVEC4-10), whereas current studies were performed using HUVEC.
DVT is considered a thromboinflammatory disease,10,70 where the production of IL-1β and activation of the endothelium are known to play significant roles in the generation of thrombus.16,41 Our in vitro studies in endothelial cells revealed that MALT1-dependent signaling might regulate many key cellular events that contribute to thromboses, such as mobilization of P-selectin to the endothelial cell surface, VWF secretion, and expression of VCAM1 and TF, which are collectively known to cause accumulation of leukocytes, activation of the coagulation system, and thrombus formation.4,13,38 Consistent with the in vitro findings, MALT1 inhibition by a specific pharmacological inhibitor significantly reduced the incidence of venous thrombosis and thrombus growth induced by the IVC ligation-induced stasis or stenosis. MALT1 inhibition markedly reduced the F4/80+ monocytes and Ly-6G+ neutrophil accumulation in the thrombus. It is likely that the inhibition of P-selectin expression on the cell surface, VWF secretion, and expression of VCAM1 and TF in MALT1 inhibitor-treated mice might be responsible for the reduced leukocyte accumulation and thrombus generation. Reduced concentrations of VCAM1, VWF, and TF in thrombi in mice treated with MALT1 inhibitor support the above concept.
MALT1 is expressed by many cell types, including endothelial cells, neutrophils, and monocytes,53 and all these cell types contribute to the pathogenesis of DVT.1 Therefore, despite our in vitro studies establishing that MALT1 plays a central role in Gab2-dependent thromboinflammatory signaling in endothelial cells, and the selective inhibition of MALT1 blocks the endothelial cell-derived prothrombotic mediators in both in vitro and in vivo settings, the present data do not allow us to conclude that the reduced thrombus formation in MALT1 inhibitor-treated animals is solely because of its effect on endothelial cells. Our current study shows that MALT 1 inhibition not only blocks the accumulation of neutrophils and monocytes in the thrombus but also inhibits NETosis. Several studies have shown the presence of NETs in venous thrombi in the IVC stenosis models and the contribution of NETs to the propagation of venous thrombosis.4,71-74 Therefore, it is possible that inhibition of NETosis by MALT1 inhibitor could have contributed to protection against venous thrombosis in the stenosis model. Although NETs might not be playing a major role in the stasis-induced venous thrombosis in wild-type mice,73 pieces of evidence for NETosis were observed in murine stasis models.75,76 Either depletion of neutrophils or DNAse treatment was shown to reduce thrombus size in the stasis models in Toll-like receptor 9-deficient mice75 and tumor-bearing mice,76 indicating that NETosis also contributes to the stasis-induced venous thrombosis under certain conditions. Depletion of neutrophils and inhibition of NETosis following the administration of MALT1 inhibitor, in which MALT1 inhibitor also suppresses endothelial cell activation and subsequent expression of prothrombotic mediators, may contribute to reduced thrombus size in the stasis model. The possibility that MALT1 inhibition in neutrophils and other cells could have contributed to the reduced venous thrombosis and the limitation in obtaining definitive proof that endothelial cell-specific inhibition of MALT1 is responsible for protection against venous thrombosis in the current study does not reduce in vivo biological significance of the present finding. An intervention that targets multiple inflammatory steps, including inhibition of cytokine signaling, cell adhesion receptors, TF, and NETosis, will be highly effective in decreasing thrombosis. MALT1 could be such a target.
Conclusions
Our study revealed a previously unrecognized role for the Gab2–MALT1 axis in orchestrating the prothrombotic signaling and expression of prothrombotic mediators elicited by IL-1β. Importantly, our study provides proof of the concept that targeting the Gab2–MALT1 axis using MALT1 inhibitors would be beneficial to treat thromboinflammation and thrombogenesis in DVT without inducing bleeding complications associated with direct inhibitors of coagulation. Further studies are required to address the cell-specific contribution of Gab2–MALT1 signaling in the pathogenesis of DVT.
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute HL107483 and HL124055 and endowment funds from The Dr. and Mrs. James Vaughn Professorship in Biomedical Research to L.V.M.R.
We dedicate this article to Usha R. Pendurthi, who died unexpectedly. She was crucial in developing the project and completing the work, including the manuscript preparation. We recognize her contribution to hemostasis, thrombosis, inflammation, and cancer research and her devotion to mentoring young researchers and educating high school and undergraduate students in medicine and research by starting and running a clinical and research externship program at our institute.
Authorship
Contribution: V.K. was involved in designing the study, performed all in vitro studies described in the paper, analyzed the data, and wrote an initial draft of the manuscript; S.K. performed the IVC ligation-induced stenosis and stasis studies in mice; K.D. and J.M. performed the immunohistochemistry analysis; L.V.M.R. and U.R.P. contributed to the study design, review and analysis of data, and writing the manuscript; and all authors reviewed the manuscript and contributed to the preparation of the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Usha R. Pendurthi died on 5 January 2022.
Correspondence: L. Vijaya Mohan Rao, Department of Cellular and Molecular Biology, The University of Texas Health Science Center at Tyler, Tyler, TX 75708; e-mail: vijay.rao@uthct.edu.
For original data or experimental protocols, please contact viay.rao@uthct.edu.
The full-text version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal