

In this issue of Blood, Ferrada et al1 demonstrate that patients with vacuoles, E1-enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome caused by 1 of 3 canonical UBA1 mutations (p.Met41Val) have more systemic inflammatory syndromes, worse survival, and lower residual translation of the normal cytoplasmic UBA1 isoform UBA1b. This links VEXAS pathogenesis and severity to a loss of UBA1b function.
VEXAS syndrome is a recently described adult-onset autoinflammatory syndrome caused by somatic mutations in UBA1, a ubiquitin-activating enzyme, within myeloid progenitor cells. The landmark paper describing VEXAS syndrome by Beck et al2 identified 25 male patients with somatic UBA1 mutations and common syndromic features including systemic inflammation and fevers, ear and nose chondritis, neutrophilic dermatosis, pulmonary infiltrates, myelodysplastic syndrome (MDS), and plasma cell disorders. They identified 3 canonical UBA1 variants that give rise to VEXAS: p.Met41Leu (c.121A>C), p.Met41Val (c.121A>G), and p.Met41Thr (c.122T>C). These constitute ∼95% of VEXAS-associated UBA1 variants reported in the literature.3-9 Subsequently, a cohort of 116 patients with VEXAS from France was described by Georgin-Lavialle et al,3 confirming the phenotypic features from the original publication. In the French cohort, the presence of a p.Met41Leu variant, but not p.Met41Thr, was associated with an improved survival relative to the p.Met41Val variant; p.Met41Val patients also had more systemic inflammation and less chondritis.3 Although these findings suggested that VEXAS might have heterogeneous clinical manifestations that are partially dependent on the specific UBA1 variant, this genotype-phenotype correlation had not previously been precisely defined nor the mechanism underpinning these differences understood.
The authors aimed to answer these questions by analyzing a cohort of 83 patients with VEXAS and canonical UBA1 mutations from the National Institutes of Health and Leeds Teaching Hospital. The patient characteristics were comparable to previous reports, with fever, skin involvement, arthritis, pulmonary infiltrates, and chondritis being the most common clinical features and approximately one-third having a diagnosis of MDS. As observed with the French cohort, patients carrying the p.Met41Val variant had less ear chondritis and more systemic disease with undifferentiated fever syndromes, while patients with p.Met41Thr had more ocular involvement. A novel association between p.Met41Leu and neutrophilic dermatosis (Sweet syndrome) was also observed. The authors then demonstrate a notably worse survival among VEXAS patients with the p.Met41Val variant, with no long-term survivors in this group, while showing comparatively better outcomes for those with p.Met41Leu or p.Met41Thr variants. A multivariable analysis for survival identified ear chondritis as a protective factor, whereas patients who were transfusion dependent or carried the p.Met41Val variant had a worse survival (see figure).
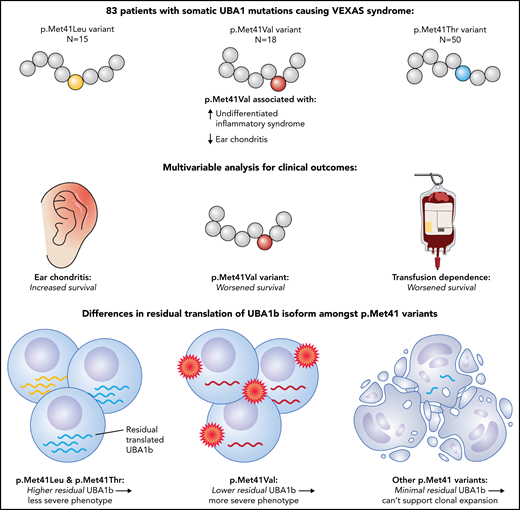
VEXAS patients with the p.Met41Val variant had more systemic undifferentiated inflammatory syndromes and less ear chondritis. A multivariable analysis for survival showed improved outcomes with ear chondritis and worse outcomes with transfusion dependence or the p.Met41Val variant. The p.Met41Leu and p.Met41Thr variants permit higher residual translation of normal cytoplasmic UBA1 (UBA1b), whereas p.Met41Val had lower residual UBA1b, and other permutations at p.Met41 had minimal residual UBA1b, linking VEXAS pathogenesis and severity to a loss of UBA1b function.
VEXAS patients with the p.Met41Val variant had more systemic undifferentiated inflammatory syndromes and less ear chondritis. A multivariable analysis for survival showed improved outcomes with ear chondritis and worse outcomes with transfusion dependence or the p.Met41Val variant. The p.Met41Leu and p.Met41Thr variants permit higher residual translation of normal cytoplasmic UBA1 (UBA1b), whereas p.Met41Val had lower residual UBA1b, and other permutations at p.Met41 had minimal residual UBA1b, linking VEXAS pathogenesis and severity to a loss of UBA1b function.
Although these differences may at first appear to be discordant with the findings from the French cohort, who identified p.Met41Leu as a marker for good prognosis and did not identify p.Met41Val as a marker for poor prognosis, it is instructive to look at the differences in follow-up between the 2 groups. Patients within the French cohort had a maximum follow-up time of 5 years, whereas most patients in the present work had between 5 and 10 years of follow-up, with several having >10 years. Within the author's cohort, no patients with p.Met41Leu died before 5 years, consistent with the French observation; indeed, the survival curves between the 2 studies appear comparable up to the 5-year mark. However, the genotype-specific impacts on outcomes become clearer after 5 to 10 years of follow-up, where the worsened survival among p.Met41Val patients becomes evident, and the survival curves for p.Met41Thr and p.Met41Leu patients converge.
The authors subsequently sought to identify the mechanism that underpins the genotype-phenotype association observed with the p.Met41Val variant using a combination of in vitro models and primary cells from VEXAS patients. Mutations at p.Met41 in UBA1 result in a loss of the translation start site for the cytoplasmic isoform (UBA1b) and use of an alternate start site (p.Met67) that produces the catalytically inactive UBA1c isoform. The presence of UBA1c does not appear to drive the phenotypic features, given that a rare UBA1 variant that does not result in UBA1c production (p.Ser56Phe) can produce a VEXAS phenotype.2,4,10 The authors use isoform-specific antibodies to show that the p.Met41Thr and p.Met41Leu variants permit some residual translation of intact UBA1b, whereas p.Met41Val produces significantly less residual UBA1b relative to the other 2 variants. The authors then perform in vitro testing on all other possible single-nucleotide variants at the p.Met41 start codon, demonstrating that these produce significantly lower levels of residual UBA1b than p.Met41Val. The authors suggest that this explains why the 3 canonical p.Met41 variants are the ones observed in VEXAS patients, with UBA1b levels lower than that seen with p.Met41Val likely being incompatible with clonal expansion and/or cell survival. Further to this, the authors describe a patient with 2 novel UBA1 variants in cis on the same allele; 1 variant (c.121A>T; p.Met41LeuTTG) reduced in vitro UBA1b translation below the minimum threshold, but this increased to similar levels as p.Met41Val upon coexpression of the second variant (p.Gly40Ala), suggesting that the occurrence of this second variant was able to partially compensate for the p.Met41LeuTTG variant.
These findings demonstrate that patients with VEXAS have genotype-specific features and that those with p.Met41Val variants seem to have a more severe clinical course and worse survival. Further, the biological basis for this appears to be a lower level of residual UBA1b translation, demonstrating an inverse relationship between UBA1b and disease severity in VEXAS, and confirming that loss of UBA1b is the primary driver of the disease phenotype. More studies are needed to address what downstream pathways lead from a decrease in UBA1b to systemic inflammation and to assess both the direct cellular effects and cell-type specificity of a loss of UBA1b function. These findings have important clinical implications for risk stratification and, potentially, therapy selection in VEXAS. Should patients with the p.Met41Val variant be selected for allogeneic hematopoietic stem cell transplantation? Are there differences in therapeutic responses (eg, azacytidine, ruxolitinib) between VEXAS genotypes? These remain important questions to be further assessed in future studies. Significantly, the identification of the centrality of the loss of UBA1b translation in VEXAS opens the possibility of future therapeutic approaches that could restore UBA1b function within cells.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal