

Key Points
Rates of subsequent malignancies in patients receiving retrovirally modified cellular therapies are similar to those in control patients.
Tumors obtained from patients with subsequent malignancies were negative for transgenes and replication competent retrovirus.

Abstract
Subsequent malignancies are well-documented complications in long-term follow-up of cancer patients. Recently, genetically modified immune effector (IE) cells have shown benefit in hematologic malignancies and are being evaluated in clinical trials for solid tumors. Although the short-term complications of IE cells are well described, there is limited literature summarizing long-term follow-up, including subsequent malignancies. We retrospectively reviewed data from 340 patients treated across 27 investigator-initiated pediatric and adult clinical trials at our center. All patients received IE cells genetically modified with γ-retroviral vectors to treat relapsed and/or refractory hematologic or solid malignancies. In a cumulative 1027 years of long-term follow-up, 13 patients (3.8%) developed another cancer with a total of 16 events (4 hematologic malignancies and 12 solid tumors). The 5-year cumulative incidence of a first subsequent malignancy in the recipients of genetically modified IE cells was 3.6% (95% confidence interval, 1.8% to 6.4%). For 11 of the 16 subsequent tumors, biopsies were available, and no sample was transgene positive by polymerase chain reaction. Replication-competent retrovirus testing of peripheral blood mononuclear cells was negative in the 13 patients with subsequent malignancies tested. Rates of subsequent malignancy were low and comparable to standard chemotherapy. These results suggest that the administration of IE cells genetically modified with γ retroviral vectors does not increase the risk for subsequent malignancy.
Introduction
Immune effector cells (IECs) genetically modified with γ retroviral vectors (GRVs) or lentiviral vectors expressing chimeric antigen receptor (CAR) T cells are used increasingly to treat patients with hematologic malignancies and are being explored in patients with solid tumors.1-7 When used in combination with lymphodepleting chemotherapy, multiple short-term toxicities of IECs have been reported. However, there is limited literature on potential long-term effects of these therapies, including their genotoxicity.8,9 Subsequent malignancies are among the reported long-term adverse events effects of many standard-of-care cancer therapies, including chemotherapy and hematopoietic stem cell transplant (HSCT), with an incidence of 2% to 5%.8,10,11 Subsequent malignancies remain a potential concern for those receiving GRV-transduced IECs, given reports of their occurrence after transfer of transduced hematopoietic stem cells for inherited diseases such as X-linked severe combined immunodeficiency.12,13 To date, there have been no reported subsequent malignancies in patients receiving GRV-transduced IECs for the treatment of hematologic malignancies or solid tumors.
In this study, we report our Center’s 30-year experience of institutionally generated IECs that were genetically modified using GRVs, representing more than 1000 years of cumulative patient follow-up. The transgenic elements in our studies include components intended to retarget the cells to tumor antigens, improve proliferative capacity, mark the IECs to track persistence, and limit apoptotic activity. We included genetically modified IEC platforms based on activated T cells or virus-specific T lymphocytes (VSTs). We followed both pediatric and adult patients, and a variety of different disease types were included, encompassing both hematologic and solid malignancies. Thirteen patients developed malignancies 1.5 to 172 months after IEC infusion, and we were unable to detect the transferred genetic element in any of the 11 tumors tested. Overall, we observed the same incidence of subsequent cancers in recipients of gene-modified cells as in high-risk patients receiving unmodified IECs.
Methods
Study design and logistics
A total of 340 adult and pediatric patients who received ≥1 GRV-transduced genetically modified IECs for a hematologic or solid tumor were retrospectively evaluated on a Baylor College of Medicine (BCM) Institutional Review Board (IRB)-approved protocol. Patients were treated with multiple lines of therapy before enrollment. Their follow-up began on study entry and was conducted by the relevant principal investigator or co-investigator as described in the study-specific calendar. If a patient was lost to follow-up or died on the study, this was recorded at the first available time. If a subsequent malignancy was suspected from clinical symptoms and signs, confirmation was made by laboratory analyses and imaging. Biopsy for transgene testing was requested from all these patients. In our assessment, relapse of the patient’s primary malignancy was not considered a subsequent malignancy. Patients were followed on 28 studies from 1 January 1993 to 4 May 2021.
Five classes of genetically modified IEC were administered to patients: (1) donor-derived gene-marked VSTs in 1 study,14 (2) autologous gene-marked VSTs in 2 studies,15 (3) donor T cells transduced with an inducible caspase 9 suicide gene in 2 studies,16,17 (4) dominant-negative tumor necrosis factor-β receptor–transduced VSTs in 3 studies,18 and (5) CAR T cells with 7 distinct target antigens in 19 studies.19-27 The packaging cell line for donor-marked (ETNA) and automarked (ANGEL/ANGELA) studies used was the PA317 packaging cell line (provided by Genetic Therapy Inc).28 For all other studies, we used the murine embryonic fibroblastic PG13 packaging cell line (obtained from ATCC; #CRL-10686) that produces gibbon ape leukemia virus GalV pseudotyped retroviral particles.29 The retroviral vector backbone for these studies was SFG and (originally provided by R. C. Mulligan, Cambridge, MA).30Table 1 lists the different constructs used in the study. All patients were treated on protocols approved by the US Food and Drug Administration and by IRBs at St Jude Children’s Research Hospital (3 studies) or BCM (27 studies, including the 3 that were also open at St Jude Children’s Research Hospital). The BCM IRB approved the current retrospective analysis.
Different types of immune effectors included in the study
| Neo marker gene | TGF-β DNR | CAR/cytokine | CAR | iC9 | |
|---|---|---|---|---|---|
| Auto-VSTs | + | + | |||
| Donor VSTs | + | + | + | ||
| Donor allodepleted ATCs | + | ||||
| Auto ATCs | + | + | + | ||
| PA317* | + | ||||
| PG13† | + | + | + | + |
| Neo marker gene | TGF-β DNR | CAR/cytokine | CAR | iC9 | |
|---|---|---|---|---|---|
| Auto-VSTs | + | + | |||
| Donor VSTs | + | + | + | ||
| Donor allodepleted ATCs | + | ||||
| Auto ATCs | + | + | + | ||
| PA317* | + | ||||
| PG13† | + | + | + | + |
allo, allogeneic; ATC, activated T cell; auto, autologous; DNR, dominant-negative receptor; iC9, inducible caspase 9.
PA317: amphitropic retrovirus packaging cell line was used for auto-VSTs and donor VSTs (obtained by Genetic Therapy Inc).
PG13: murine embryonic fibroblastic PG13 packaging cell lines (obtained from ATCC; #CRL-10686) were used for all other GRV-IEC types.
To determine whether GRV genetic modification of IECs increases the risk of subsequent malignancies, we included a control group of patients who received IECs that had not been genetically modified before infusion. This group included 111 patients with Hodgkin lymphoma (HL) or B-cell non-Hodgkin lymphoma (NHL) who were treated with Epstein-Barr VSTs (EBVSTs). Like the genetically modified cell group, patients were evaluated for time to second malignancy starting at enrollment.
GRV transgene and replication-competent retrovirus testing
GRV transgenes were detected via quantitative polymerase chain reaction (PCR) amplification of peripheral blood mononuclear cells (PBMCs) collected preinfusion and at standardized times after IEC infusion as per the study calendar. Transgene copy number in the infused cell lines and the follow-up blood samples of patients on protocols with genetically modified T cells are measured by quantitative PCR (qPCR), using TaqMan primers and probes custom designed to detect the specific transgene in genomic DNA extracted from the IEC product and patients’ PBMC as previously described for the individual studies.14-28 The National Gene Vector Biorepository at Indiana University (Indianapolis, IN) tested samples for replication competent retrovirus (RCR) in PBMCs preinfusion and subsequently as required by the study-specific calendar.
Outcome and statistical design
Descriptive statistics were used to summarize patient characteristics.31 Cumulative follow-up was used to describe the total duration of follow-up for all patients on the treatment group by summing all the individual follow-up years of each patient. The primary endpoint of this study was the presence of a subsequent malignancy. Time to subsequent malignancy was recorded from the time of the first infusion on the study to the time of the first diagnosis of subsequent disease. The cumulative incidence of these malignancies was estimated and compared between genetically modified IECs and genetically unmodified modified IECs by the competing risk method as described by Gray.32 The subdistribution hazard ratios were estimated and compared between the treatment groups using the Fine-Gray subdistribution hazard regression model.33 Rates are calculated with exact 95% binomial confidence intervals (CIs). All P values are 2-sided, and values of P < .05 were considered statistically significant.34
Results
Patient characteristics
A total of 340 patients received GRV genetically modified IECs. Of these, 204 (60.0%) were adult and 136 (40.08%) were pediatric patients with a median age at enrollment of 23.5 years (range, 1-78). One hundred and eighty-eight (55.3%) patients had primary hematologic malignancy, 150 (44.1%) had solid tumors, and 2 (0.6%) had nonmalignant conditions. The most common hematologic malignancy was NHL, followed by HL and acute lymphoblastic leukemia. Among solid tumors, osteosarcoma and neuroblastoma were the most common (Table 2). Of the 340 patients, 99 (29.1%) had autologous HSCT and 83 (24.4%) had an allogeneic HSCT. The median follow-up of patients was 14.9 months (range, >1-183), with 192 (56.5%), 72 (21.2%), and 29 (8.5%) patients followed for more than 1, 5, and 10 years, respectively.
Baseline patient characteristics
| Characteristic | All patients, n = 340 |
|---|---|
| Age, median (range), y | 23.5 (1-78) |
| Sex, n (%) | |
| Male | 197 (57.9) |
| Female | 143 (42.1) |
| Primary diagnosis, n (%) | |
| Hematologic malignancy | 188 (55.3) |
| Solid malignancy | 150 (44.1) |
| Nonmalignant conditions | 2 (0.6) |
| Hematologic malignancies, n (%) | |
| ALL | 46 (24.5) |
| AML | 11 (5.9) |
| CLL | 11 (5.9) |
| CML | 8 (4.3) |
| NHL | 41 (21.8) |
| HL | 54 (28.7) |
| MM | 7 (3.7) |
| Others | 10 (5.3) |
| Solid tumors, n (%) | |
| Neuroblastoma | 33 (22.0) |
| Glioblastoma | 19 (12.7) |
| Lung cancer | 10 (6.7) |
| Osteosarcoma | 52 (34.7) |
| Head and neck | 16 (10.7) |
| Liver tumors | 8 (5.3) |
| Others | 12 (8.0) |
| Transplant, n (%) | |
| Autologous | 99 (29.1) |
| Allogeneic | 83 (24.4) |
| No transplant | 158 (46.5) |
| History of radiation therapy, n = 339 (%) | |
| No | 137 (40.4) |
| Yes | 202 (59.6) |
| Median follow-up (range), mo | 14.9 (<1-183) |
| Cumulative follow-up, y | 1027 |
| Characteristic | All patients, n = 340 |
|---|---|
| Age, median (range), y | 23.5 (1-78) |
| Sex, n (%) | |
| Male | 197 (57.9) |
| Female | 143 (42.1) |
| Primary diagnosis, n (%) | |
| Hematologic malignancy | 188 (55.3) |
| Solid malignancy | 150 (44.1) |
| Nonmalignant conditions | 2 (0.6) |
| Hematologic malignancies, n (%) | |
| ALL | 46 (24.5) |
| AML | 11 (5.9) |
| CLL | 11 (5.9) |
| CML | 8 (4.3) |
| NHL | 41 (21.8) |
| HL | 54 (28.7) |
| MM | 7 (3.7) |
| Others | 10 (5.3) |
| Solid tumors, n (%) | |
| Neuroblastoma | 33 (22.0) |
| Glioblastoma | 19 (12.7) |
| Lung cancer | 10 (6.7) |
| Osteosarcoma | 52 (34.7) |
| Head and neck | 16 (10.7) |
| Liver tumors | 8 (5.3) |
| Others | 12 (8.0) |
| Transplant, n (%) | |
| Autologous | 99 (29.1) |
| Allogeneic | 83 (24.4) |
| No transplant | 158 (46.5) |
| History of radiation therapy, n = 339 (%) | |
| No | 137 (40.4) |
| Yes | 202 (59.6) |
| Median follow-up (range), mo | 14.9 (<1-183) |
| Cumulative follow-up, y | 1027 |
ALL, acute lymphocytic leukemia; AML, acute myelocytic leukemia; CML, chronic myelocytic leukemia; MM, multiple myeloma; TBI, total body irradiation.
The control group consisted of 111 patients treated with genetically unmodified EBVSTs. Their median age was 44 (range, 6-78). All patients had lymphoma, 51 (45.9%) with HL and 60 (54.1%) with B-cell NHL. Of the 111 patients, 37 (33.3%) had autologous HSCT and 13 (11.7%) had an allogeneic HSCT (Table 3).
Control group baseline characteristics
| Characteristic | All patients, n = 111 |
|---|---|
| Age, median (range), y | 44 (6-78) |
| Sex, n (%) | |
| Male | 73 (65.8) |
| Female | 38 (34.2) |
| Primary diagnosis (%) | |
| Hodgkin lymphoma | 51 (45.9) |
| Non-Hodgkin lymphoma | 60 (54.1) |
| Transplant, n (%) | |
| Autologous | 37 (33.3) |
| Allogeneic | 13 (11.7) |
| No transplant | 61 (55.0) |
| Included protocols | GRALE (NCT01555892) ALCI and ALASCER (NCT00062868) |
| Characteristic | All patients, n = 111 |
|---|---|
| Age, median (range), y | 44 (6-78) |
| Sex, n (%) | |
| Male | 73 (65.8) |
| Female | 38 (34.2) |
| Primary diagnosis (%) | |
| Hodgkin lymphoma | 51 (45.9) |
| Non-Hodgkin lymphoma | 60 (54.1) |
| Transplant, n (%) | |
| Autologous | 37 (33.3) |
| Allogeneic | 13 (11.7) |
| No transplant | 61 (55.0) |
| Included protocols | GRALE (NCT01555892) ALCI and ALASCER (NCT00062868) |
General characteristics in patients with subsequent malignancies
Of the 340 patients treated with a cumulative follow-up of 1027 years, 13 (3.8%; 95% CI, 2.1-6.4) patients developed a total of 16 subsequent malignancies (Table 4). Three of the 13 (23%) patients had more than 1 subsequent malignancy. The median age of all patients with subsequent malignancies at enrollment was 27 (range, 6-62). Ten of the 13 patients were males. Twelve of 16 (75%) subsequent malignancies were solid tumors, the most common being basal cell carcinoma, with 4/16 (25%) total malignancies. There was a broad range of tumors diagnosed within this solid tumor subgroup with no site favored (Table 4). Subsequent malignancies occurred 1.5 to 172 months (median, 45 months) after IEC infusion, with 10 (62.5%) malignancies occurring within 5 years and 6 (38%) malignancies occurring more than 10 years after IEC therapy. Of the 13 patients, 8 were still alive at the last follow-up. Subsequent malignancy contributed to death in 2 patients, although 5 died with subsequent malignancies.
Characteristics of patients that developed subsequent malignancies after genetically modified IECs
| Patient | Age at enrollment (y) | Sex | Primary disease | HSCT | IEC type | Subsequent neoplasm | Time from IEC therapy, mo | Tissue transgene detection | RCR | Time to LFU, y | Outcome at last follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6 | M | Neuroblastoma | Yes (auto-HSCT) | GD2 CAR T | AML | 1.5 | Negative | Negative | 3 | Alive |
| 2 | 27 | F | HL | Yes (auto-HSCT) | DNR TGF-β EBVSTs | MDS | 24 | Negative | Negative | 3 | Died of complications after allogeneic transplant |
| 3 | 17 | M | Nasopharyngeal carcinoma | No | DNR TGF-β EBVSTs | Spindle-cell neoplasm | 24 | Negative | Negative | 4.5 | Alive |
| 4 | 62 | M | CLL | Yes (allo-HSCT) | CD19 CAR T | Bladder cancer | 48 | Negative | Negative | 5 | Died of secondary infection |
| 5 | 11 | M | Glioblastoma | No | HER2 CAR T | T-cell lymphoma | 36 | Negative | Negative | 6 | Died of T-cell lymphoma |
| 6 | 60 | M | CLL | No | CD19 CAR T | BCCx2* | 36 and 48 | Unable to obtain | Negative | 6 | Alive |
| 7 | 47 | F | HL | Yes (auto-HSCT) | DNR TGF-β EBVSTs | Breast ductal cell carcinoma | 42 | Negative | Negative | 7 | Alive |
| 8 | 55 | M | NHL | Yes (auto-HSCT) | CD19 CAR T | BCC | 36 | Unable to obtain | Negative | 7.5 | Alive |
| 9 | 56 | M | NHL | No | CD19 and κ CAR T | 1-Penile squamous cell carcinoma | 72† | Negative | Negative | 7.5 | Died of MDS complications |
| 2-MDS | 36 | Negative | Negative | ||||||||
| 10 | 36 | M | HL | Yes (auto-HSCT) | Auto EBVSTs with NeoR | 1-RCC | 154 | Unable to obtain | Negative | 15 | Alive |
| 2-Meningioma | 172 | Unable to obtain | Unable to obtain | ||||||||
| 11 | 18 | M | ALL and secondary AML | Yes (allo-HSCT) | Allo EBVSTs with NeoR | Nerve sheath tumor‡ | 96 | Negative | Negative | 15 | Died of unrelated accident |
| 12 | 8 | M | AML | Yes (allo-HSCT) | Allo EBVSTs with NeoR | Thyroid adenoma | 144 | Unable to obtain | Negative | 15 | Alive |
| 13 | 15 | F | ALL | Yes (allo-HSCT) | Allo EBVSTs with NeoR | BCC | 156 | Negative | Negative | 15 | Alive |
| Patient | Age at enrollment (y) | Sex | Primary disease | HSCT | IEC type | Subsequent neoplasm | Time from IEC therapy, mo | Tissue transgene detection | RCR | Time to LFU, y | Outcome at last follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6 | M | Neuroblastoma | Yes (auto-HSCT) | GD2 CAR T | AML | 1.5 | Negative | Negative | 3 | Alive |
| 2 | 27 | F | HL | Yes (auto-HSCT) | DNR TGF-β EBVSTs | MDS | 24 | Negative | Negative | 3 | Died of complications after allogeneic transplant |
| 3 | 17 | M | Nasopharyngeal carcinoma | No | DNR TGF-β EBVSTs | Spindle-cell neoplasm | 24 | Negative | Negative | 4.5 | Alive |
| 4 | 62 | M | CLL | Yes (allo-HSCT) | CD19 CAR T | Bladder cancer | 48 | Negative | Negative | 5 | Died of secondary infection |
| 5 | 11 | M | Glioblastoma | No | HER2 CAR T | T-cell lymphoma | 36 | Negative | Negative | 6 | Died of T-cell lymphoma |
| 6 | 60 | M | CLL | No | CD19 CAR T | BCCx2* | 36 and 48 | Unable to obtain | Negative | 6 | Alive |
| 7 | 47 | F | HL | Yes (auto-HSCT) | DNR TGF-β EBVSTs | Breast ductal cell carcinoma | 42 | Negative | Negative | 7 | Alive |
| 8 | 55 | M | NHL | Yes (auto-HSCT) | CD19 CAR T | BCC | 36 | Unable to obtain | Negative | 7.5 | Alive |
| 9 | 56 | M | NHL | No | CD19 and κ CAR T | 1-Penile squamous cell carcinoma | 72† | Negative | Negative | 7.5 | Died of MDS complications |
| 2-MDS | 36 | Negative | Negative | ||||||||
| 10 | 36 | M | HL | Yes (auto-HSCT) | Auto EBVSTs with NeoR | 1-RCC | 154 | Unable to obtain | Negative | 15 | Alive |
| 2-Meningioma | 172 | Unable to obtain | Unable to obtain | ||||||||
| 11 | 18 | M | ALL and secondary AML | Yes (allo-HSCT) | Allo EBVSTs with NeoR | Nerve sheath tumor‡ | 96 | Negative | Negative | 15 | Died of unrelated accident |
| 12 | 8 | M | AML | Yes (allo-HSCT) | Allo EBVSTs with NeoR | Thyroid adenoma | 144 | Unable to obtain | Negative | 15 | Alive |
| 13 | 15 | F | ALL | Yes (allo-HSCT) | Allo EBVSTs with NeoR | BCC | 156 | Negative | Negative | 15 | Alive |
ALL, acute lymphocytic leukemia; Allo, allogeneic; AML, acute myelocytic leukemia; auto, autologous; BCC, basal cell carcinoma; CLL, chronic lymphoid leukemia; LFU, last follow-up; MDS, myelodysplastic syndrome; NeoR, neomycin resistant marker gene; RCC, renal cell carcinoma; RCR, replication-competent retrovirus; TGF, transforming growth factor.
Patient was diagnosed with 2 basal cell carcinomas of the skin at 36 and 48 mo after infusion.
Patient received both CD-19 and κ CAR. Time to last follow-up and to secondary malignancy was calculated from time of his first infusion (CD19 CAR).
Patient with history of neurofibromatosis.
Cumulative incidence of subsequent malignancies in gene-modified IEC recipients
We compared the cumulative incidence of the first subsequent malignancy among patients receiving genetically modified IECs group vs patients treated with genetically unmodified EBVSTs. Of 340 patients treated, 13 (3.8%) developed subsequent malignancies. A total of 195 (57.4%) died and 132 (38.8%) were alive at the last follow-up (up to 15.2 years) without subsequent malignancies. We observed that the 5-year cumulative incidence of the first subsequent malignancy in the recipients of genetically modified cells was 3.6% (95% CI, 1.8-6.4). Of the 111 patients receiving genetically unmodified cells, 4 patients (3.6%) developed subsequent malignancies (2 myelodysplastic syndrome, 2 sarcoma) at 5 years, whereas 39 (35.1%) died and 68 (61.3%) were alive at the last follow-up (up to 5 years) without subsequent malignancies. The 5-year cumulative incidence of subsequent malignancy in this population was 4.2% (95% CI, 1.4-9.7) (Figure 1). Thus, at 5 years, there was no difference in the cumulative incidence of the first subsequent malignancy between the 2 groups (P = .953, Gray test). The estimated subdistribution hazard ratio of modified cells compared with unmodified was 0.9 (95% CI, 0.30-2.9), and thus there was no significant effect for the cumulative incidence of subsequent malignancies (P = .859 by the Fine-Gray subdistribution hazard regression model).
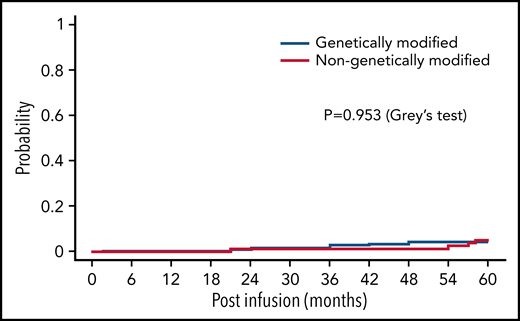
Cumulative incidence of subsequent malignancies. Cumulative incidence plots showing incidence of γ retroviral genetically modified IECs vs genetically unmodified IECs.
Cumulative incidence of subsequent malignancies. Cumulative incidence plots showing incidence of γ retroviral genetically modified IECs vs genetically unmodified IECs.
Distribution of subsequent malignancies across studies with genetic modification
Neither the source of IEC nor the transgene itself discernibly influenced the risk of subsequent malignancies. Of the 16 subsequent malignancies reported, 3 occurred in patients receiving donor gene-marked IECs, 2 in recipients of autologous gene-marked IECs, 3 in recipients of IECs expressing a dominant negative receptor for tumor necrosis factor-β, and 8 in patients treated with CAR T cells. Patients on 3 additional CAR T-cell studies had no subsequent malignancies (CD30, glypican 3, and CD5); although the CD30 study opened 10 years ago, the glypican-3 and CD5 studies started enrolling patients in the past 5 years (Table 5).
Details of included clinical trials
| Type of modification | Type of immune effector cells | Trial names (cumulative follow-up, y) | Trials included (NCI no.) | No. of patients | No. of SMs | Median time to last detected transgene, mo (range)* |
|---|---|---|---|---|---|---|
| Donor marked | EBVSTs | ETNA (287.6) | NCT00058812 | 26 | 3 | 44.5 (0-105) |
| Auto marked | EBVSTs | ANGEL and ANGELA (19.1) | NCT00058617 | 7 | 2 | Up to 2 y |
| iCasp9 | iCasp9 T cell | CASPALLO (48.3) DOTTI (28.0) | NCT00710892 NCT01494103 | 24 | 0 | 12 (0.25-108) |
| Dominant negative receptor | DNR TGF-β cells | TGFB (38.1) RESIST NPC (17.9) HESTIA (13.3) | NCT00675571 NCT02065362 NCT02379520 | 28 | 3 | 6 (0.5-72) |
| CAR T cell | GD2 CAR T cells | NESTLES (93.1) GRAIN (30.0) GAIL-N (10.3) VEGAS (16.2) | NCT00085930 NCT01822652 NCT03635632 NCT01953900 NCT03294954 | 51 | 1 | 3 (0-108) |
| CD19 CAR T cells | CRETI (59.2) ATECRAB (8.5) SAGAN (47.0) CARSPASCIO (15.5) MULTIPRAT (55.6) | NCT00586391 NCT00709033 NCT01853631 NCT02050347 NCT00840853 | 63 | 6 | 6 (0-96) | |
| HER2 CAR T cells | HERCREEM (20.6) HERT-GBM (15.9) HEROS (71.1) | NCT00889954 NCT01109095 NCT00902044 | 71 | 1 | 1.5 (0-84) | |
| CD30 CAR T cells | CAR CD30 (18.6) CART CD30 (30.3) RELY 30 (26.0) | NCT01192464 NCT01316146 NCT02917083 | 33 | 0 | 6 (0-24) | |
| Glypican 3 CAR T cells | GLYCAR (1.8) GAP (3.6) | NCT02905188 NCT02932956 | 8 | 0 | 3 (2-12) | |
| CD5 CAR T cells | MAGENTA (6.7) | NCT03081910 | 11 | 0 | 3 (0.75-30) | |
| κ CAR T cells | CHARKALL (45.1) | NCT00881920 | 18 | 2† | 6 (0.25-60) |
| Type of modification | Type of immune effector cells | Trial names (cumulative follow-up, y) | Trials included (NCI no.) | No. of patients | No. of SMs | Median time to last detected transgene, mo (range)* |
|---|---|---|---|---|---|---|
| Donor marked | EBVSTs | ETNA (287.6) | NCT00058812 | 26 | 3 | 44.5 (0-105) |
| Auto marked | EBVSTs | ANGEL and ANGELA (19.1) | NCT00058617 | 7 | 2 | Up to 2 y |
| iCasp9 | iCasp9 T cell | CASPALLO (48.3) DOTTI (28.0) | NCT00710892 NCT01494103 | 24 | 0 | 12 (0.25-108) |
| Dominant negative receptor | DNR TGF-β cells | TGFB (38.1) RESIST NPC (17.9) HESTIA (13.3) | NCT00675571 NCT02065362 NCT02379520 | 28 | 3 | 6 (0.5-72) |
| CAR T cell | GD2 CAR T cells | NESTLES (93.1) GRAIN (30.0) GAIL-N (10.3) VEGAS (16.2) | NCT00085930 NCT01822652 NCT03635632 NCT01953900 NCT03294954 | 51 | 1 | 3 (0-108) |
| CD19 CAR T cells | CRETI (59.2) ATECRAB (8.5) SAGAN (47.0) CARSPASCIO (15.5) MULTIPRAT (55.6) | NCT00586391 NCT00709033 NCT01853631 NCT02050347 NCT00840853 | 63 | 6 | 6 (0-96) | |
| HER2 CAR T cells | HERCREEM (20.6) HERT-GBM (15.9) HEROS (71.1) | NCT00889954 NCT01109095 NCT00902044 | 71 | 1 | 1.5 (0-84) | |
| CD30 CAR T cells | CAR CD30 (18.6) CART CD30 (30.3) RELY 30 (26.0) | NCT01192464 NCT01316146 NCT02917083 | 33 | 0 | 6 (0-24) | |
| Glypican 3 CAR T cells | GLYCAR (1.8) GAP (3.6) | NCT02905188 NCT02932956 | 8 | 0 | 3 (2-12) | |
| CD5 CAR T cells | MAGENTA (6.7) | NCT03081910 | 11 | 0 | 3 (0.75-30) | |
| κ CAR T cells | CHARKALL (45.1) | NCT00881920 | 18 | 2† | 6 (0.25-60) |
DNR, dominant-negative receptor; NCI, National Cancer Institute; TGF, transforming growth factor.
Time to last detected transgene was calculated from the time of last infusion (if patient received >1 infusion) to time to any positive transgene value detected by qPCR.
Two subsequent malignancies were reported in a patient who received both CD19 and κ CAR T cells.
Transgene detection and RCR testing
To determine whether gene-modified cells directly contributed to subsequent malignancies, we used qPCR to measure transgene prevalence in peripheral blood and tumors. All 13 subsequent malignancy patients had RCR testing of peripheral blood at multiple time points after IEC therapy and no patient had RCR detectable in PBMCs before or after malignancy onset. Table 5 includes IEC and study-specific details on transgene persistence. Additionally, 11 of 13 patients with subsequent malignancy had tumor biopsies available. We did not detect any IEC transgenes in any of these 11 biopsies.
Although 1 patient developed T-cell lymphoma 2 years following HER2–CAR T-cell therapy, he was previously diagnosed with constitutional mismatch repair deficiency syndrome, an aggressive cancer predisposition syndrome with mediastinal T-cell lymphoma being the most common.35,36 No IEC transgene was detected in the lymphoma biopsy, and RCR testing was negative at all time points before and after infusion.
Discussion
We reviewed 340 patients who received ≥1 GRV-modified IEC with a cumulative follow-up of 1027 years. To do date, this is the longest follow-up of patients receiving retroviral genetically modified IECs. Our cohort of patients included a wide range of ages (range, 1-78 years), as well as varied primary diagnoses, encompassing solid tumors, malignant hematologic diseases, and nonmalignant immunodeficiencies. In this retrospective analysis, we demonstrate no increased risk of subsequent malignancy in patients treated with retroviral genetically modified IECs. Additionally, all patients treated on this study had exposure to chemotherapy before enrollment on each respective clinical trial. We show that the risk of subsequent malignancy in this patient group is 3.6%, which is similar to the 2% to 5% range that has been documented in previous literature for patients treated with chemotherapy.7
In this study, RCR testing was negative in all patients at all time points, and no IEC transgenes were detected in any tumor biopsies form patients with subsequent malignancies after IEC infusion. These findings reinforce Bear et al’s study on RCR detection, in which they looked at 27 clinical studies using genetically modified T-cell products with GRVs and did not detect any RCR in 42 viral supernatant lots or any T-cell products and Cornetta et al’s evaluation of 241 patients over 14 clinical trials, none of whom had detectable RCR infection.37,38
Although retroviral genetically modified IECs do not appear to increase the risk of subsequent malignancy in our study, other factors which contribute to malignant transformation and necessitate close follow-up. For example, Brown et al examined whether CD40 ligand (CD40L) deficiency, known as X-linked hyper-IgM syndrome, could be corrected via gene therapy. The authors transduced murine bone marrow or thymic cells with a retroviral vector containing complementary DNA for murine CD40 ligand and injected them into deficient mice. Although treatment led to improved immune response in mice, 12 of 19 mice developed T-cell lymphomas. Their data suggested that retroviral genetic modification did not lead to malignant transformation because RCR was not detected in samples, nor were retroviral particles identified by electron microscopic examination in tumors. Furthermore, insertional mutagenesis seemed unlikely because proviral DNA was only noted in a small group of tumor cells. Instead, subsequent malignancies were thought to be secondary to CD40L’s role in thymocyte regulation via upregulation of costimulatory molecules on thymocyte accessory cells and naïve T lymphocytes. Thus, the constitutive expression of CD40L in immature thymocytes potentially lead to a malignant T-cell clone because the CD40L transgene was detected in tumor tissue in these mice.39
Although no lentiviral genetically modified IECs have been associated with secondary malignancies to date, clonal expansion from insertional mutagenesis and subsequent malignancies remain a risk. Shah et al reported 1 patient presenting with asymptomatic leukocytosis around 50 days after receiving treatment with lentiviral genetically modified CD22-CAR T cells for relapsed B-cell ALL. A dominant T-cell clone containing a copy of the lentiviral vector integrated into the intron of the CBL gene was detected. Following treatment with steroids the patient was reevaluated at day +180, and the clone was not detected.40 Similarly, Fraietta et al reported a case of clonal CAR T-cell expansion in the context of anti-CD19 CAR T-cell therapy for a 70 year old with relapsed chronic lymphocytic leukemia. This was likely because of a concomitant lentiviral integration into the TET2 gene, along with a hypomorphic mutation on the patient’s second allele. Because TET2 plays a role in regulating hematopoiesis and T-cell differentiation, this integration likely led to the patient’s CAR T-cell expansion and antitumor effect.41 These cases of clonal expansion demonstrate that lentiviral integration into host genes pose a risk of insertional mutagenesis, prompting a need for continued long-term follow-up.
Other genetic transfer systems, such as the piggyBac transposon system, offer significant advantages to viral vectors because of lower cost and ability to transfer large genetic inserts42,43; however, a theoretical risk of genomic rearrangement and malignant transformation remains. Bishop et al used CD19-CAR T cells generated with the piggyBac transposon system to treat 10 CD19+ leukemia and lymphoma patients. Although treatment appeared effective, 2 of 10 patients developed secondary monoclonal CAR T-cell malignancies 12 months and 16 months after treatment. CAR T expression was noted in biopsies of both patients, indicating malignant transformation; neither malignancy had integration of the CAR transgene into oncogenes, and there was no unique driver of malignant transformation for the 2 patients. Bishop et al note that their production protocol, which included electroporation and varying concentrations of transposon and transposase, were likely factors in the malignant transformation of these patients.44 Of note, Gregory et al recently treated 23 patients using a piggyBac transposon generated CAR T cells targeting B-cell maturation antigen. There were no significant toxicities noted, nor were there subsequent malignancies at 3 years posttherapy.45,46
Additionally, gene-editing strategies including CRISPR-Cas9 and transcription activator-like effector nucleases have been used to modify CAR T cells to reduce the risk of graft-versus-host disease or rejection.47 Transcription activator-like effector nuclease disruption of the TRAC gene and the CD52 locus was recently associated with a chromosomal abnormality on chromosome 14, the location of the TRAC gene, but in a region associated with myeloproliferative disorders. Although there was clonal expansion of the modified cells, no progression to malignancy has occurred to date.48
In an initial analysis of 48 of our patients receiving GRV-modified cells, we did not see an association between malignant transformation and vector copy number. We observed a mean vector copy number per transduced cell of 5.38 (range, 1.48-9.40). That said, most studies for which vector copy number was calculated are within the first 5 years of follow-up; thus, more long-term assessment is necessary.44
The 1 patient on our study who developed a mediastinal T-cell lymphoma after HER2-CAR T-cell infusion had no evidence of the CAR T transgene in the tumor biopsy sample nor in the peripheral blood. Additionally, he was known to have constitutional mismatch repair deficiency, which put him at increased risk for hematologic malignancies, with mediastinal lymphoma being particularly prevalent in patients with this deficiency. There is no evidence to date of subsequent resulting from GRV modification in T cells, as demonstrated by multiple groups.49,50
In summary, our data reinforce the safety of retroviral genetically modified IECs, and in our series show that GRV-modified vectors did not significantly increase the risk of subsequent malignancy in patients. Additional long-term studies will be needed to further demonstrate the safety and risk of subsequent malignancies in the transposon systems and in lentiviral IECs.
Acknowledgments
The authors thank all the physicians who cared for these patients, the Good Manufacturing Practice (GMP) facility, Good Laboratory Practice (GLP) facility, and clinical research staff who participated in these studies and the patients for participating in long-term follow up.
This work was supported by grants from the National Institutes of Health, National Cancer Institute (P50CA126752, 1U54CA232568-01, and P01CA094237), Stand Up To Cancer (SU2C)/St Baldrick’s Pediatric Cancer Dream Team Translational Research Grant (SU2C-AACR-DT1113), Stand Up To Cancer (SU2C)/American Association for Cancer Research (AACR) 604817 Meg Vosburg T-Cell Lymphoma Dream Team, and the Leukemia and Lymphoma Society. SU2C is a program of the Entertainment Industry Foundation administered by the AACR.
Authorship
Contribution: D.H.M.S. and I.N.M. drafted the manuscript; T.W. and M.W. performed statistical analyses; H.E.H., C.A.R., L.C.H., D.H.M.S., and I.N.M. designed the study; D.H.M.S. and I.N.M. performed data collection; N.A., M.H., S.B., O.D., S.G., S.B.W., P.D.L., M.M., B.O., R.H.R., A.H., L.S.M., L.H., C.A.R., C.M.R., M.K.B., and H.E.H. contributed to products manufacturing, samples analysis, patient enrollment, and edited the manuscript; and all authors approved the final version of the manuscript.
Disclosures
M.K.B. and C.M.R. have equity in Allovir, Marker Therapeutics, and Tessa Therapeutics; have served on advisory boards for Walking Fish Therapeutics, CellGenix GmbH, Marker Therapeutics, Tessa Therapeutics, Abintus, Allogene, Bellicum Pharmaceuticals, Bluebird Bio, Athenex, Memgen, Turnstone Biologics, Coya Therapeutics, TScan Therapeutics, Onkimmune, Poseida Therapeutics, and Allovir; and have received research support from Tessa Therapeutics. H.E.H. has equity in Allovir and Marker Therapeutics; has served on advisory boards for Tessa Therapeutics, Novartis, Gilead, GSK, Kiadis, and Fresh Wind Biotechnologies; and received research support from Tessa Therapeutics and Kuur Therapeutics. S.G. has a consulting agreement with Tessa Therapeutics; is a compensated DSMB member of Immatics; has received honoraria from Tidal, Catamaran Bio, and Novartis within the last 2 years; and has received research support from Tessa Therapeutics in the past. P.L. served on an advisory board for Karyopharm Therapeutics. B.G. owns QBRegulatory Consulting, which has agreements with LOKON Pharma, TESSA Therapeutics, Marker Therapeutics, and AlloVir. L.M. and A.H. report research support from Athenex, Inc.
Correspondence: David Steffin, Center for Cell and Gene Therapy, Baylor College of Medicine/Texas Children's Hospital, Feigin Center, 1102 Bates St, Ste 730.05, Houston, TX 77030; e-mail: dhsteffi@texaschildrens.org.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal