

In this issue of Blood, report the discovery of novel pathogenic variants in replication protein A1 (RPA1, encoded by RPA1) as a new cause of telomere biology disorders (TBDs).1 This highly effective multi-institutional collaboration characterized the clinical manifestations in 4 individuals from 4 families, uncovered biology suggesting a role for RPA1 in hematopoiesis, built upon understanding of RPA1’s role in telomeric DNA binding and unfolding, and discovered the somatic rescue of the mutation in a patient. This discovery advances understanding of RPA function in telomere biology and, importantly, helps families understand the cause of their illness and their long diagnostic journey and sets the stage for further advances in understanding the role of telomere biology and DNA repair in human disease.
TBDs represent a spectrum of clinical phenotypes united by a common underlying biology.2,3 The first TBD, X-linked dyskeratosis congenita (DC), is caused by germline mutations in dyskerin (DKC1) and is classically diagnosed by the mucocutaneous triad of oral leukoplakia, nail dystrophy, and abnormal skin pigmentation. TBDs are also associated with high rates of bone marrow failure, myelodysplastic syndrome (MDS), acute myeloid leukemia, oral squamous cell carcinoma, and/or pulmonary fibrosis. Implementation of the diagnostic test, lymphocyte telomere lengths less than the first percentile for age measured by flow cytometry with in situ hybridization (flow FISH),4 and rapid advances in genomics have led to discoveries of variants in more than a dozen genes (TERT, TERC, TINF2, NOP10, NHP2, WRAP53, RTEL1, PARN, ACD, POT1, CTC1, STN1, NAF1, and ZCCHC8) with autosomal dominant and/or recessive inheritance contributing to TBD etiology (see figure).2,3 The frequent occurrence of variable expressivity and penetrance resulting in variable phenotypes has led to designation of such illnesses as TBDs because the term reflects a common biology and is not dependent on a defined telomere length, which can be variable, particularly in adult-onset heterozygous disease.4-6
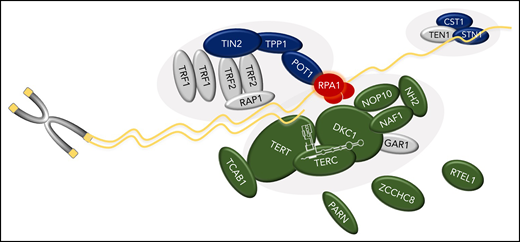
Schematic of RPA1’s role in telomere biology and relationship to other genes associated with TBD. RPA1, the largest subunit of the RPA heterotrimeric complex, binds single-stranded DNA (ssDNA) at telomeres. TBD-associated variants in RPA1 result in increased ssDNA binding affinity. The components of the telomerase enzyme complex (DKC1, TERC, TERT, NAF1, NOP10, NHP2), telomerase or hTR regulators (TCAB1, PARN, and ZCCHC8), and regulator of telomere elongation helicase 1 (RTEL1), are shown in green. Dark blue indicates the shelterin (TPP1, TIN2, and POT1) and CST (CTC1 and STN1) complexes. Protein names are shown. Gray symbols indicate known key telomere biology proteins not yet attributed to human disease (GAR1 in telomerase complex; TRF1, TRF2, and RAP1 in shelterin; and TEN1 in CST).
Schematic of RPA1’s role in telomere biology and relationship to other genes associated with TBD. RPA1, the largest subunit of the RPA heterotrimeric complex, binds single-stranded DNA (ssDNA) at telomeres. TBD-associated variants in RPA1 result in increased ssDNA binding affinity. The components of the telomerase enzyme complex (DKC1, TERC, TERT, NAF1, NOP10, NHP2), telomerase or hTR regulators (TCAB1, PARN, and ZCCHC8), and regulator of telomere elongation helicase 1 (RTEL1), are shown in green. Dark blue indicates the shelterin (TPP1, TIN2, and POT1) and CST (CTC1 and STN1) complexes. Protein names are shown. Gray symbols indicate known key telomere biology proteins not yet attributed to human disease (GAR1 in telomerase complex; TRF1, TRF2, and RAP1 in shelterin; and TEN1 in CST).
The clinical manifestations in patients reported by Sharma et al are consistent with TBDs and illustrate their diagnostic complexities. Patient 1 (RPA1 c.718G>A, p.E240K) had pancytopenia at age 10 years and developed the DC mucocutaneous triad in adolescence. Patient 2 (RPA1 c.680T>C, p.V227A) had MDS and a hematopoietic cell transplantation complicated by pulmonary fibrosis, infection, and graft vs host disease. Patient 3, from an unrelated family, had the same RPA1 variant and developed pulmonary fibrosis; two of her siblings passed away from complications of pulmonary fibrosis. Patient 1’s persistent hyperplastic primary vitreous as well as patient 2 and her carrier father’s dysmorphic eye findings are notable for not being typical TBD features. Patient 4 (c.808A>G, p.T270A) had lymphopenia and hypogammaglobulinemia, features sometimes, but not always, present in TBDs. Patient 1’s lymphocyte telomeres, measured by flow FISH at less than the first percentile for age, were consistent with her clinical presentation. Patient 4’s flow FISH lymphocyte telomeres started out technically normal at the fifth percentile but shortened to slightly less than the first percentile in a year and a half. Telomeres for patients 2 and 3 were measured by Southern blot and telomere shortest length assay (TeSLA) and notable for being much shorter than healthy controls.
Telomere protection and maintenance requires precise interactions between DNA replication and repair proteins, telomeric DNA folding, extension of telomeres by telomerase, and protection of telomeres by the shelterin and CTC1/STN1/TEN1 (CST) complexes.7 The heterotrimeric RPA complex binds to ssDNA to prevent formation of secondary structures and to regulate DNA binding by other proteins. Mutations in RPA have been postulated but not previously shown to contribute to TBDs in humans; studies in yeast and mouse models showed aberrant DNA damage repair and chromosomal instability associated with RPA1 mutations.8,9
Sharma et al provide the first evidence that missense variants in RPA1 contribute to TBD etiology. Each of the affected amino acids (p.E240K, p.V227A, and p.T270A) have PhastCons scores of 1, suggesting high degrees of evolutionary conservation, but variable phyloP scores (7.64, 4.81, and 1.14, respectively). These key differences appear to also be reflected in the functional data. Both p.E240K and p.V227A mutant heterodimers act as gain-of-function (GOF) mutants as demonstrated by increased binding affinity to ssDNA and telomeric sequences and increased rates of telomere unfolding. In contrast, p.T270A’s ssDNA and telomeric DNA binding affinity was only marginally increased. Further evaluation in induced pluripotent stem cells homozygous for patient 1’s p.E240K GOF mutation found shorter telomeres and lower yields of erythroid and myeloid populations compared with controls. Given that her cytopenias did not progress as would have been expected during 20 years of follow-up, Sharma et al evaluated whether acquisition of hematopoietic somatic mutations could have resulted in somatic hematopoietic reversion. Patient 1’s primary germline RPA1 variant was present at an allele fraction of only 27% in her bone marrow but at 50% in her fibroblasts. Over 15 years, the germline variant (c.718G>A, p.E240K) declined as the somatic variant increased (c.1745G>T, p.K579*).
The addition of pathogenic variants in RPA1 to the family of TBD-associated genes is an opportunity for the field to expand understanding of the intricate links between telomere maintenance and the DNA damage response (see figure). The patient phenotypes described by Sharma et al further illustrate the importance of being open to unexpected diagnoses, to connect family history with disease (whenever possible), and to conduct detailed longitudinal follow-up. It is impossible to overstate the value of open collaborative studies through programs such as the Undiagnosed Disease Network.10
Key questions remain in understanding RPA biology in the context of human disease including (1) Should RPA1 be added to bone marrow failure gene panels? (2) Is fibroblast mutation testing required for all (or some) marrow failure syndromes and, if so, should it be combined with somatic testing? (3) How common are RPA1 pathogenic variants in TBDs, and are there genotype-phenotype associations? Answering these questions is possible, even in rare causes of rare diseases, provided the clinical and basic science communities continue to pursue international collaborations across phenotypes and biologic pathways and remain open to new discoveries that may or may not connect one hypothesis with another.
Conflict-of-interest disclosure: S.A.S. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal