

In this issue of Blood, Ruben et al1 determine structures of the prothrombinase complex, both bound to lipid nanodiscs and in a ternary complex with prothrombin. This study illustrates, for the first time, how factors Va (fVa) and Xa (fXa) associate on a membrane surface, as well as how prothrombin binds in a conformation that leads to the generation of meizothrombin.
Hemostasis is maintained by a series of proteolytic events that convert inactive zymogens into active serine proteases at the sight of vascular injury, ultimately leading to the formation of an insoluble fibrin mesh.2 The penultimate step in the blood coagulation cascade is the proteolytic conversion of prothrombin to thrombin by the prothrombinase complex,3 consisting of fVa and fXa in the presence of calcium and negatively charged phospholipids on the membrane surface of activated platelets or endothelial cells.
Activation of prothrombin is a complex process that has been characterized in detail, yet no direct structural information has been reported.3 The complete generation of thrombin requires 2 site-specific cleavages of prothrombin, at R271 and R320, which is described via 2 distinct pathways.4 Recent studies have shown that the prothrombinase complex prefers initial cleavage at R320, generating the catalytically active meizothrombin, followed by cleavage at R271 to release mature thrombin, which serves to cleave fibrinogen to initiate the formation of the insoluble fibrin mesh.5 The alternative pathway results from initial cleavage at R271, generating the inactive intermediate prothrombin-2, followed by subsequent cleavage at R320 to generate mature thrombin. The structural features of prothrombin have been recalcitrant to structural studies owing to conformational heterogeneity. Recent single-molecule FRET experiments illustrated that prothrombin exists in an equilibrium between an “open” and “closed” state.6 Using site-directed mutagenesis, constructs of prothrombin have locked both conformations, yielding zymogen states that were amenable to structure determination by X-ray crystallography.7,8 Further studies have demonstrated that meizothrombin is generated from cleavage in the “closed” conformation, inducing movement to the open conformation for secondary R271 cleavage.5
The prothrombinase complex has also lacked structural characterization; however, several models have been proposed based on complementary studies. Although there have been several structures determined for thrombin and fXa, which have led to the development of next-generation anticoagulants that are direct inhibitors of these serine proteases, the factor V (fV)/fVa complete structures have only recently been determined with cryogenic electron microscopy (cryo-EM).9 FVa plays a critical cofactor role in the prothrombinase complex, increasing the catalytic activity of fXa for prothrombin by >1000-fold.
fV circulates as an inactive protein procofactor consisting of an A1-A2-B-A3-C1-C2 domain architecture. Through activation by thrombin, fVa is specifically cleaved at R709, R1018, and R1545. Activation releases the unconserved B domain that keeps fV in an inactive state. The nascently generated C-terminus of the A2 domain (residues 654-709) has been shown to impact prothrombinase assembly and catalytic activity, but this region is largely undefined in the fVa structure. The cofactor function played by fVa is to associate fXa and prothrombin in an optimal arrangement whereby Xa can site-specifically cleave prothrombin, initially at R320, on a negatively charged membrane surface. Membrane association for each component of the prothrombinase ternary complex is achieved either through the C domains (for FVa) or through γ-carboxyglutamic acid (Gla) domains (for prothrombin and FXa). Ruben et al have for the first time determined the structure of prothrombinase bound to lipid nanodiscs as well as in a ternary complex with prothrombin in its “closed” state, in a conformation that illustrates how R320 is oriented toward the fXa active site, leading to the generation of meizothrombin.
The lipid nanodisc-bound structure, which represents a membrane-bound assembly, was determined to lower resolution (5.3 Å). The structure illustrates how fVa associates with membrane surfaces through its C domains and supports previous models for membrane association.10 The majority of the prothrombinase complex occurs between the fVa A2 domain and the protease domain of fXa. Intrinsically disordered in the previously determined fVa structure, the 672 to 691 segment of fVa A2 domain C-terminus forms a lidlike cover over the protease domain of fXa, representing a conformational shift from the fV structure by >7 Å. Other electrostatic and hydrophobic interactions also occur between the fVa A2 domain and the protease domain of fXa, presumably contributing to the formation of a prothrombinase-active conformational state. Minor contacts are also observed between the A3 and EGF2 domains, as well as the C1 and Gla domains, of fVa and fXa, respectively (see upper figure).
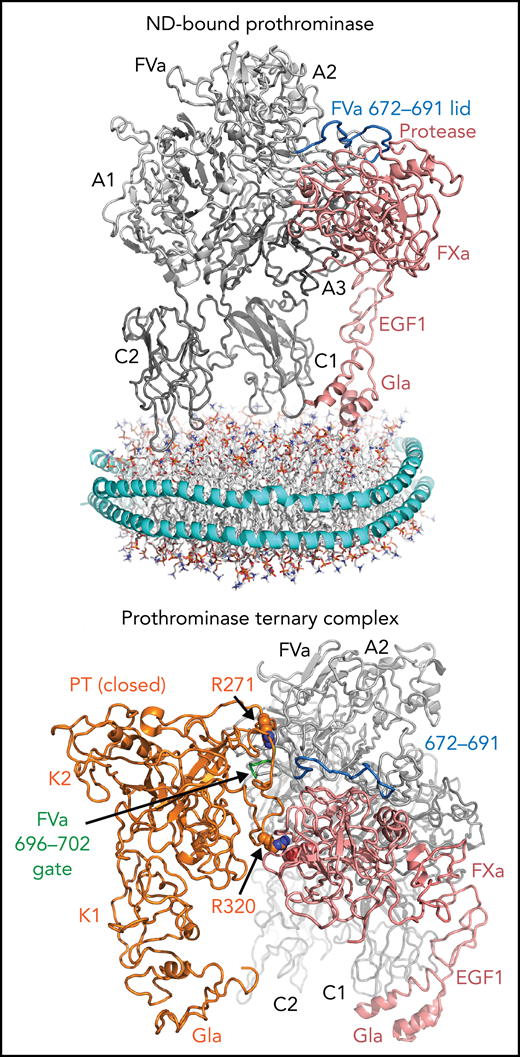
Cryo-EM structures of lipid nanodisc-bound prothrombinase (top) and prothrombinase in a ternary complex with prothrombin (bottom). Top, FVa is represented in gray, with the A1, A2, A3, C1, and C2 domains labeled. FXa is shown in pink with the Gla, EGF1, and protease domains labeled, forming connections with the FVa A2, A3, and C1 domains. The protease domain of FXa is held in place with an FVa A2 domain “lid” (residues 672-691, shown in blue). The lipid nanodisc is modeled below to illustrate how these domains bind membrane surfaces. Bottom, prothrombinase (FVa/FXa) bound to prothrombin in its “closed” form. Prothrombin is shown in orange, with the Gla and Kringle (K1 and K2) domains labeled, which makes a small contact with the FVa A2 domain 696 to 702 loop (green). R320 projects toward the specificity site adjacent to the active site of FXa (pink). R271 sits above the 696 to 702 loop. EGF1, epidermal growth factor-like domain 1; ND, lipid nanodisc; PT, prothrombin.
Cryo-EM structures of lipid nanodisc-bound prothrombinase (top) and prothrombinase in a ternary complex with prothrombin (bottom). Top, FVa is represented in gray, with the A1, A2, A3, C1, and C2 domains labeled. FXa is shown in pink with the Gla, EGF1, and protease domains labeled, forming connections with the FVa A2, A3, and C1 domains. The protease domain of FXa is held in place with an FVa A2 domain “lid” (residues 672-691, shown in blue). The lipid nanodisc is modeled below to illustrate how these domains bind membrane surfaces. Bottom, prothrombinase (FVa/FXa) bound to prothrombin in its “closed” form. Prothrombin is shown in orange, with the Gla and Kringle (K1 and K2) domains labeled, which makes a small contact with the FVa A2 domain 696 to 702 loop (green). R320 projects toward the specificity site adjacent to the active site of FXa (pink). R271 sits above the 696 to 702 loop. EGF1, epidermal growth factor-like domain 1; ND, lipid nanodisc; PT, prothrombin.
Remarkably, the prothrombinase structure is conserved in a ternary complex with prothrombin in the absence of lipid nanodiscs, strongly suggesting this is an authentic, physiologically relevant assembly. The long segment of the A2 domain C-terminus, 680 to 709, was previously implicated in prothrombin binding and prothrombinase activity; however, only a small portion (696-702) serves to separate the 2 sites of cleavage. Here, this is proposed to induce directionality in prothrombin cleavage, sequestering R271 away from the fXa active site while directing R320 immediately for proteolysis. Surprisingly, only modest interactions occur between the protease domain of prothrombin with the A2 domain of fVa. Several previous contacts reported through indirect measurements were not supported by the prothrombinase ternary complex determined in this study, including no direct interactions of the Kringle or Gla domains of prothrombin with either fVa or fXa. The overall structure of the ternary complex is compatible with all membrane association domains being presented along the same plane to bind phospholipid surfaces simultaneously, forming a domelike structure (see lower figure).
The 2 structures reported by Ruben et al represent a landmark advancement in the understanding of how thrombin is activated and provides an optimistic route toward successfully elucidating the structures of more membrane-associated coagulation complexes in the future. Lipid nanodisc technology, single-molecule FRET measurements, and cryo-EM advances can all now be combined to generate great new insights into how blood coagulation occurs rapidly and specifically at the sites of vascular damage and may lead to more innovative clinical interventions to improve treatments for thrombotic and hemophilic diseases.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal