

Key Points
Selective inhibition of APC anticoagulant activity prevents joint bleed–induced HA.
Endogenous APC cytoprotective effects play a crucial role in preserving joint health in hemophilia.

Abstract
Recurrent spontaneous or trauma-related bleeding into joints in hemophilia leads to hemophilic arthropathy (HA), a debilitating joint disease. Treatment of HA consists of preventing joint bleeding by clotting factor replacement, and in extreme cases, orthopedic surgery. We recently showed that administration of endothelial cell protein C receptor (EPCR) blocking monoclonal antibodies (mAb) markedly reduced the severity of HA in factor VIII (FVIII)−/− mice. EPCR blocking inhibits activated protein C (APC) generation and EPCR-dependent APC signaling. The present study was aimed to define the role of inhibition of APC anticoagulant activity, APC signaling, or both in suppressing HA. FVIII−/− mice were treated with a single dose of isotype control mAb, MPC1609 mAb, that inhibits anticoagulant, and signaling properties of APC, or MAPC1591 mAb that only blocks the anticoagulant activity of APC. Joint bleeding was induced by needle puncture injury. HA was evaluated by monitoring joint bleeding, change in joint diameter, and histopathological analysis of joint tissue sections for synovial hypertrophy, macrophage infiltration, neoangiogenesis, cartilage degeneration, and chondrocyte apoptosis. No significant differences were observed between MPC1609 and MAPC1591 in inhibiting APC anticoagulant activity in vitro and equally effective in correcting acute bleeding induced by the saphenous vein incision in FVIII−/− mice. Administration of MAPC1591, and not MPC1609, markedly reduced the severity of HA. MAPC1591 inhibited joint bleed–induced inflammatory cytokine interleukin-6 expression and vascular leakage in joints, whereas MPC1609 had no significant effect. Our data show that an mAb that selectively inhibits APC’s anticoagulant activity without compromising its cytoprotective signaling offers a therapeutic potential alternative to treat HA.
Introduction
Chronic joint bleeding in patients with hemophilia results in hemophilic arthropathy (HA), a degenerative joint disease with a remarkable negative impact on mobility and quality of life.1-3 HA is characterized by synovitis comprising synovial hyperplasia, infiltration of inflammatory cells, neoangiogenesis in the synovial space, articular cartilage, and subchondral bone damage.4-8 Despite the widespread availability of safe and effective replacement therapies, many patients with hemophilia continue to develop the joint disease because of breakthrough bleeding and the development of inhibitors.7,9,10 Recombinant factor VIIa (FVIIa) or activated prothrombin complex concentrates (APCCs) were commonly used as bypassing agents for patients with hemophilia with inhibitors.11
Research over the past 2 decades led to the development of nontraditional agents, whose mechanisms of action differ from current bypassing agents, for treating patients with hemophilia with or without inhibitors. They include the development of bispecific antibodies (emicizumab) that mimic FVIII12,13 or agents that inhibit endogenous anticoagulants, such as antithrombin,14,15 tissue factor pathway inhibitor,16,17 or activated protein C.18,19 Although nonfactor therapies appear to improve hemostasis in patients with hemophilia in clinical settings or experimental models, and emicizumab (Hemlibra) was approved for the treatment of hemophilia A patients with or without inhibitors, they may not completely prevent breakthrough bleeding and have other limitations.20-22
Interest in inhibition of activated protein C (APC) as a potential therapy in treating hemophilia stems from the observation that patients with hemophilia with factor V–Leiden mutation, which confers partial resistance to the cleavage by APC, were found to have reduced severity of bleeding.23 Selective inhibition of APC by bioengineered serpin, KRK-α-antitrypsin, was shown to rescue hemostasis in a hemophilia mouse model.18 Targeting APC with a monoclonal antibody (mAb) that specifically binds APC but not protein C and inhibits APC anticoagulant activity but not cytoprotective functions of APC was found to normalize hemostasis in monkeys with hemophilia.24 Our recent studies showed that administration of endothelial cell protein C receptor (EPCR) blocking mAbs markedly reduces recurrent joint bleeding and hemophilic synovitis in FVIII−/− mice subjected to a needle puncture injury of joints.8 EPCR blocking antibody blocks the binding of both protein C and APC to the EPCR and thus prevents generation of APC as well as APC-mediated cytoprotective signaling.25,26 Because EPCR binds other ligands and could affect various cellular processes independent of APC,27 it was difficult to conclude from the earlier study8 whether the protective effect of EPCR antibody in HA comes from the rebalancing of hemostasis through downregulation of APC generation or other mechanisms. Furthermore, it also raises a question about the importance of preserving APC-mediated cytoprotective signaling to the safe treatment of patients with hemophilia with APC targets. The present study was performed to investigate the effectiveness of APC antibodies that either selectively inhibit APC anticoagulant activity or both APC anticoagulant activity and signaling function in suppressing the development of needle puncture–induced HA in hemophilia A mice.
The data presented in the current paper show that inhibition of endogenous APC anticoagulant activity selectively with an exosite-binding mAb protects against HA. The selective inhibition of APC anticoagulant activity markedly reduced joint bleed–induced synovial inflammation, macrophage infiltration, neoangiogenesis, articular cartilage degeneration, and chondrocyte apoptosis. A mAb that inhibits both anticoagulant activity and signaling function of APC failed to protect against HA, indicating endogenous APC signaling may play a protective role in the pathogenesis of HA.
Materials and methods
Reagents
The generation and characterization of mAbs against murine protein C (MPC1609) and APC (MAPC1591) were described earlier.28 Recombinant mouse APC (MAPC) was produced in Esmon’s laboratory using standard procedures.29 Human FVa was obtained from Haemtech (Essex Junction, VT). Human factor Xa and prothrombin were obtained from Enzyme Research Laboratories (South Bend, IN).
Animals
Wild-type (WT) mice (C57BL/6J) and FVIII−/− mice were bred in-house. FVIII−/− mice in the B6/129S (Jackson Laboratories) were backcrossed with C57BL/6J mice for >10 generations to generate FVIII−/− mice in the C57BL/6J genetic background.
Neutralization of APC anticoagulant activity
Inhibition of APC anticoagulant activity was assessed in thrombin generation assay. Briefly, MAPC (20 pM) was incubated with varying dilutions of MPC1609 or MAPC1591 (0 to 200 nM) in the presence of phospholipids (phosphatidylcholine (PC):phosphatidylserine (PS):phosphatidylethanolamine (PE) in the ratio of 40:20:40; 25 µM). After 1 hour, FVa (20 pM) was added to the above incubation mixture and allowed to incubate for 30 minutes at room temperature. At the end of 30 minutes, factor Xa (0.4 nM) and prothrombin (1 µM) were added to the reaction mixture, and thrombin generated in the reaction mixtures was measured using a chromogenic substrate.
Effect of MPC1609 and MAPC1591 on APC-mediated cytoprotective effect
MAPC (50 nM) was incubated with MPC1609, MAPC1591, or control IgG (200 µg/mL) for 1 hour at 37°C, and then the mixture was added to murine brain endothelial cells. After 1 hour, endothelial cells were stimulated with interleukin-1β (IL-1β; 10 ng/mL) for 6 hours, and the cytokine IL-6 levels in the supernatant-conditioned media were measured by enzyme-linked immunosorbent assay (ELISA).
Treatments and induction of joint bleeding by needle puncture
FVIII−/− mice were administered with MPC1609, MAPC1591, or control IgG MCO1716 (1 mg kg−1 body weight) intraperitoneally (ip). After 24 hours, intraarticular bleeding into knee joints was induced by a needle puncture injury as described earlier.30,31 Briefly, the joint capsule of the right knee of anesthetized mice was punctured with a 30 × 0.5-g needle below the patella to induce bleeding into the joint. The uninjured left knee of the same mouse served as a control.
Evaluation of hemarthrosis
To assess the initial bleeding following the needle puncture, uninjured and injured knees were excised from mice 5 hours after the needle puncture. Hemoglobin levels in joint tissues were determined as described recently by us.8 Knee joint diameter, before the injury and every alternate day for 2 weeks following the injury, was measured using electronic calipers. The knee joint diameter before the injury was subtracted from the diameter following the injury, and the differences in the diameter were plotted as the percent change in the joint diameter. Every alternate day gross examination of knee joints was performed to assess the extent of joint bleeding and knee mobility visually, as described in our recent publication.8 The visual bleeding score (VBS) was evaluated in a blinded fashion where the person assigning the score was unaware of the treatments.
Histology of knee joints and synovitis scoring
For histological examination, knee joints (the femur and tibia/fibula area, ∼1 cm each direction from the joint) were collected into EXCEL buffered formalin fixative. After 48 hours of fixation, knee joints were decalcified for 16 to 20 hours, processed in graded alcohol, and embedded in paraffin. Five-micrometer thin sections were cut, and the sections were stained with hematoxylin and eosin (H&E). The H&E-stained sections were viewed under the microscope and scored for hemophilic synovitis (0 to 11 range) as described in our earlier publication.8 The knee joint tissue sections were also stained with Alcian blue and safranin-O fast green stains for proteoglycans and glycosaminoglycans to evaluate cartilage degeneration. To measure iron deposits in the injured synovium, the joint sections were stained with Prussian blue stain to detect tissue iron.
Immunohistochemistry
To evaluate the infiltration of macrophages into the synovium, joint tissue sections were stained for F4/80 antigen, and the macrophage score was assigned based on a 0 to 4 scale (no macrophages, 0; scattered macrophages, 1; lines of macrophages, 2; clusters of macrophages, 3; and sheets of macrophages, 4). Neoangiogenesis was assessed by staining knee joint tissue sections with CD31 antibodies. To quantify neoangiogenesis, the number of blood vessels was counted in multiple ×40 magnification fields covering the entire synovium and then averaged to the number of blood vessels per field. The number of apoptotic chondrocytes was estimated by staining the knee joint sections with a TUNEL stain. The number of apoptotic chondrocytes was counted in multiple fields (×40 magnification) and averaged per field.
Imaging
Tissue sections were viewed under an Olympus microscope equipped with 4×/0.13, 10×/0.30, 20×/0.50, 40×/0.75 objective lenses. Images were captured using an Olympus DP27 camera using Olympus Cellsens software.
Assessment of vascular leakage
Vascular leakage into knee joints was quantified by extravasation of fluorescein dextran as described in our previous publication.32
Collection of synovial fluid and IL-6 assay
On day 7 following the knee injury, synovial fluid from both uninjured and injured knees was collected as described in our recent publication.8 IL-6 levels in the synovial fluid were estimated using the IL-6 ELISA kit (Affymetrix) according to the manufacturer’s instructions.
Saphenous vein incision-induced bleeding
Acute bleeding was induced by the incision of the saphenous vein, as described earlier.33,34
Data analysis
Statistical significance among the groups was analyzed by either one-way or two-way analysis of variance, repeated measures followed by Tukey’s post hoc multiple comparison tests, as appropriate. When comparing 2 groups, the Mann-Whitney U test was used to determine statistical significance. Survival curves were plotted using the Kaplan-Meier method.
Results
Both MPC1609 and MAPC1591 inhibit APC anticoagulant activity, but only MPC1609 and not MAPC1591 inhibits APC cytoprotective function
MPC1609 was obtained by screening the ability of mAbs to block the binding of MPC to murine endothelial cells (bEnd3 cells) that express EPCR by FACS.28 MPC1609 was shown to inhibit protein C and APC binding to endothelial cells.28 MAPC1591 was obtained by screening the ability of mAbs to bind immobilized MAPC but not protein C in an ELISA.28 MAPC1591 binds APC and not protein C, and enhances APC binding to endothelial cells.28 Earlier studies showed that both MPC1609 and MAPC1591, which may bind different epitopes on APC, inhibit APC anticoagulant activity completely in the one‐stage coagulation assay initiated by factor X coagulant protein from Russell viper venom.28 MPC1609, and not MAPC1591, prevented cytoprotective functions of APC.28 Consistent with the earlier finding, both MPC1609 and MAPC1591 showed a dose-dependent inhibition of APC anticoagulant activity as assessed by the inhibition of APC inactivation of FVa in a prothrombinase assay (Figure 1A). At lower concentrations, MPC1609 was twofold more effective than MAPC1591 in inhibiting APC anticoagulant activity. At near saturating concentrations, no significant differences were observed between the 2 antibodies in inhibiting APC anticoagulant activity. MPC1609 inhibited ∼92% of APC anticoagulant activity, whereas MAPC1591 blocked 87% of the anticoagulant activity (Figure 1A).
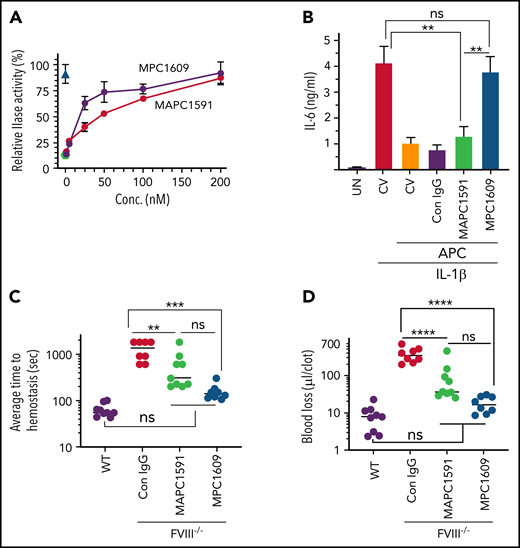
Effect of MPC1609 and MAPC1591 on the anticoagulant and cytoprotective activities of APC. (A) Varying concentrations of MPC1609 and MAPC1591 were incubated with murine APC (20 pM) and phospholipids (25 µM) for 1 hour. Then, FVa (20 pM) was added to the APC and phospholipid mixture and incubated for 30 minutes. APC inactivation of FVa was measured in prothrombinase (IIase) assay by adding factor Xa (0.4 nM) and prothrombin (1 µM) and determining thrombin generation in a chromogenic assay. IIase activity measured in the absence of APC or the antibodies and not subjected to any preincubation was taken as 100%. Green filled circle indicates relative the IIase activity in a reaction mixture containing APC but no antibodies. The blue triangle indicates relative IIase activity in the absence of both APC and antibodies. (B) Confluent monolayers of bEND3 endothelial cells were treated with control vehicle (CV), MAPC (50 nM), or MAPC (50 nM) plus control IgG, MPC1609, or MAPC1591 (200 µg/mL) for 1 hour. After 1 hour, the endothelial cells were stimulated with IL-1β (10 ng/mL) for 16 hours. At the end of 16 hours, supernatant media were collected and IL-6 levels in the media were determined by ELISA. UN, untreated; first CV (red bar) indicates cells treated with control vehicle alone; the second CV (orange bar) indicates cells treated with a CV, followed by APC. (C-D) FVIII−/− mice were administered with control IgG, MAPC1591, or MPC1609 via the tail vein (1 mg/kg). After 1 hour, the mice were subjected to the saphenous vein incision, and the average time to achieve hemostasis (C) and the blood loss (D) was determined as described in “supplemental Methods” (8 to 9 mice per group). The data shown for WT mice for comparative purposes were from the earlier study of the authors’ laboratory.34 ns, not a statistically significant difference. **P < .01; ***P <.001; ****P < .0001. conc., concentration.
Effect of MPC1609 and MAPC1591 on the anticoagulant and cytoprotective activities of APC. (A) Varying concentrations of MPC1609 and MAPC1591 were incubated with murine APC (20 pM) and phospholipids (25 µM) for 1 hour. Then, FVa (20 pM) was added to the APC and phospholipid mixture and incubated for 30 minutes. APC inactivation of FVa was measured in prothrombinase (IIase) assay by adding factor Xa (0.4 nM) and prothrombin (1 µM) and determining thrombin generation in a chromogenic assay. IIase activity measured in the absence of APC or the antibodies and not subjected to any preincubation was taken as 100%. Green filled circle indicates relative the IIase activity in a reaction mixture containing APC but no antibodies. The blue triangle indicates relative IIase activity in the absence of both APC and antibodies. (B) Confluent monolayers of bEND3 endothelial cells were treated with control vehicle (CV), MAPC (50 nM), or MAPC (50 nM) plus control IgG, MPC1609, or MAPC1591 (200 µg/mL) for 1 hour. After 1 hour, the endothelial cells were stimulated with IL-1β (10 ng/mL) for 16 hours. At the end of 16 hours, supernatant media were collected and IL-6 levels in the media were determined by ELISA. UN, untreated; first CV (red bar) indicates cells treated with control vehicle alone; the second CV (orange bar) indicates cells treated with a CV, followed by APC. (C-D) FVIII−/− mice were administered with control IgG, MAPC1591, or MPC1609 via the tail vein (1 mg/kg). After 1 hour, the mice were subjected to the saphenous vein incision, and the average time to achieve hemostasis (C) and the blood loss (D) was determined as described in “supplemental Methods” (8 to 9 mice per group). The data shown for WT mice for comparative purposes were from the earlier study of the authors’ laboratory.34 ns, not a statistically significant difference. **P < .01; ***P <.001; ****P < .0001. conc., concentration.
When the antibodies were analyzed for their ability to inhibit APC anti-inflammatory effect in endothelial cells, where it is primarily mediated through the EPCR-PAR1 axis, MPC1609 fully attenuated APC’s anti-inflammatory effect as measured by its ability to suppress IL-1β–induced expression of IL-6. In contrast, MAPC1591 did not interfere with APC-mediated suppression of IL-1β–induced IL-6 production in endothelial cells (Figure 1B). Although APC was shown to exert its cytoprotective effects primarily through the EPCR-PAR1 axis,35,36 APC may also activate other PAR receptors, such as PAR237 and PAR3.38-40 Furthermore, APC could induce cytoprotective signaling in monocytic cells, independent of EPCR and PAR1, by ligating to ApoER2.41 In endothelial cells, ApoER2 could contribute to APC’s signaling mediated through the EPCR-PAR1 axis.42 Therefore, we next investigated the effect of MPC1609 and MAPC1591 on APC activation of other PARs and ApoER2-dependent signaling. In endothelial cells transfected with PAR reporter constructs, APC is found to cleave PAR1, PAR2, and PAR3, but not PAR4 (supplemental Figure 1, available on the Blood Web site). Preincubation of MPC1609 with APC completely blocked APC’s ability to cleave PAR1, PAR2, and PAR3. MAPC1591 treatment had no significant effect on APC cleavage of PAR1, PAR2, or PAR3 (supplemental Figure 1). As reported earlier in U937 cells,41 APC-induced cytoprotective effect in murine bone marrow–derived macrophages appears to be dependent on ApoER2 and independent of EPCR, as EPCR blocking antibodies failed to block the protective effect of APC in inhibiting hemoglobin- or lipopolysaccharide (LPS)-induced expression of IL-6, whereas receptor-associated protein (RAP) that inhibits APC binding to ApoER2 attenuated APC’s protective effect (supplemental Figure 2). Neither MPC1609 nor MAPC1591 had a significant effect on the ApoER2-dependent cytoprotective effect of APC in bone marrow-derived macrophages (supplemental Figure 2). In endothelial cells and synovial fibroblasts, APC-induced cytoprotective effect appears to depend on both EPCR and ApoER2 as EPCR blocking antibodies or RAP inhibited APC’s cytoprotective activity significantly but not completely. Treatment of the cells with both EPCR blocking antibody and RAP reversed APC’s protective effect completely (supplemental Figure 3). MPC1609 treatment fully reversed APC’s cytoprotective effect in endothelial cells and synovial fibroblasts, whereas MAPC1591 had no significant effect (supplemental Figure 3).
In additional studies, we investigated the effect of MAPC1609 and MAPC1591 in restoring hemostasis in mice with hemophilia in an acute bleeding model. FVIII−/− mice were administered with a control IgG, MAPC1591, or MPC1609 (1 mg/kg) via the tail vein. One hour later, bleeding was induced by the saphenous vein incision. Both MAPC1591 and MPC1609 treatments corrected bleeding in FVIII−/− mice (Figure 1C-D). The minor differences between them in restoring hemostasis were not statistically significant. Although the average time to achieve hemostasis and the blood loss in MAPC1591- and MPC1609-treated mice were slightly higher than those observed in WT mice, the differences among them were not statistically significant (Figure 1C-D).
Next, we investigated the effect of MAPC1609 and MAPC1591 on APC activity levels and thrombin generation in blood collected at the wound site following the saphenous vein incision. In uninjured mice, trace levels of APC activity were found in the circulating blood of both WT and FVIII−/− mice. APC activity levels in blood collected at the wound site were significantly higher in WT mice compared with preinjury plasma samples (supplemental Figure 4). In contrast, APC levels were increased only marginally at the wound site in FVIII−/− mice. Administration of MPC1609 or MAPC1591 further reduced low levels of APC activity in FVIII−/− mice, but the decrease was not statistically significant (supplemental Figure 4). Measurement of thrombin generated at the wound site, as measured by thrombin-antithrombin complexes, revealed that administration of either MPC1609 or MAPC1591 significantly increased thrombin generation at the wound site in FVIII−/− mice (supplemental Figure 5). As reported earlier with recombinant FVIIa administration,43 thrombin-antithrombin levels were not significantly increased in systemic circulation following the injury or the administration of APC antibodies (supplemental Figure 5).
Assessment of initial joint bleeding in FVIII−/− mice administered with MPC1609 and MAPC1591 following the needle puncture
FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg, ip). Twenty-four hours later, mice were subjected to the needle puncture injury in knee joints. Bleeding into the joint tissue was evaluated 5 hours following the injury. Needle puncture resulted in massive joint bleeding in FVIII−/− mice. As described in our earlier report,8 the needle puncture, in addition to inducing bleeding in the joint, also resulted in massive bleeding into the soft tissue around the joint (Figure 2A). Quantification of hemoglobin extracted from the joint and adjacent soft tissue showed an equal amount of joint bleeding in FVIII−/− mice administered with control IgG, MPC1609, or MAPC1591 (Figure 2B). Measurement of hematocrit and hemoglobin content in peripheral blood showed a marked decrease in their levels in all 3 groups of mice following the joint injury. The extent of decrease in the hematocrit or hemoglobin levels was not statistically significant among the 3 groups (Figure 2C-D).
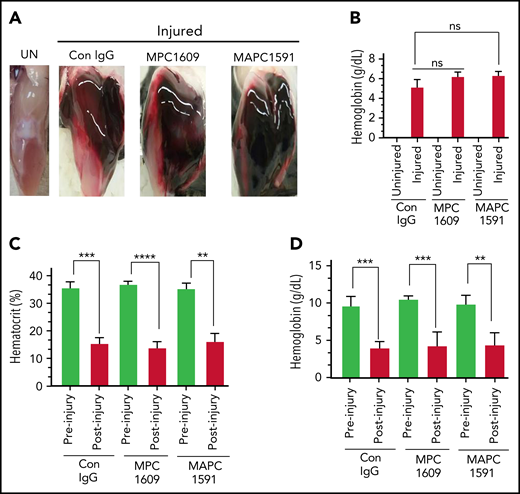
No differences in initial joint bleeding in FVIII−/− mice administered with MPC1609 and MAPC1591 following the needle puncture of the joint. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg) ip. Twenty-four hours later, the right knee joint capsule of the anesthetized mouse was punctured with a 30 × 0.5-g needle below the patella to induce bleeding into the joint. The left knee joint of the same animal served as uninjured control (UN). Five hours following the injury, the mice were euthanized, and the skin was removed over the knee joints. The knee joints were photographed, and joint tissues were processed to extract hemoglobin. Blood samples were collected from the submandibular vein puncture before euthanasia to measure hematocrit and hemoglobin content. (A) Representative photographs of uninjured and injured knees; (B) hemoglobin content extracted from joint tissues of uninjured and injured knees; (C) hematocrit; (D) hemoglobin levels in peripheral blood samples. ns, not statistically significant difference. **P < .01; ***P < .001; ****P < .0001 (n = 4 mice per group).
No differences in initial joint bleeding in FVIII−/− mice administered with MPC1609 and MAPC1591 following the needle puncture of the joint. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg) ip. Twenty-four hours later, the right knee joint capsule of the anesthetized mouse was punctured with a 30 × 0.5-g needle below the patella to induce bleeding into the joint. The left knee joint of the same animal served as uninjured control (UN). Five hours following the injury, the mice were euthanized, and the skin was removed over the knee joints. The knee joints were photographed, and joint tissues were processed to extract hemoglobin. Blood samples were collected from the submandibular vein puncture before euthanasia to measure hematocrit and hemoglobin content. (A) Representative photographs of uninjured and injured knees; (B) hemoglobin content extracted from joint tissues of uninjured and injured knees; (C) hematocrit; (D) hemoglobin levels in peripheral blood samples. ns, not statistically significant difference. **P < .01; ***P < .001; ****P < .0001 (n = 4 mice per group).
MAPC1591 that blocks APC anticoagulant activity but not APC signaling reduces joint edema following the needle puncture–induced joint bleeding
Measurement of knee joint diameter before the injury and following the injury for 14 days showed that the joint diameter was increased by ∼50% to 60% on day 2 following the injury in FVIII−/− mice administered with control IgG or MPC1609 (Figure 3A). The joint edema was decreased slightly but not statistically significant from day 2 to day 14. No significant differences were observed in joint edema between mice administered with control IgG or MPC1609. The increase in the joint diameter was significantly lower in FVIII−/− mice that received MAPC1591 than mice administered with control IgG or MPC1609 at all time points (Figure 3A).
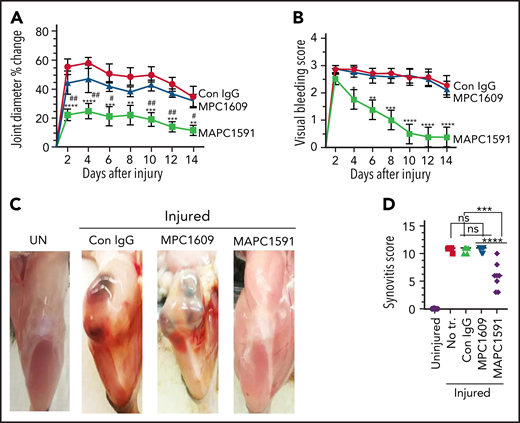
Administration of MAPC1591, and not MPC1609, reduces joint bleeding, joint edema, and synovitis in hemophilia A mice following needle puncture joint injury. A single dose of control IgG, MPC1609, or MAPC1591 (1 mg/kg) was administered to FVIII−/− mice ip. Twenty-four hours later, joint bleeding was induced with a needle puncture. Mice were monitored for 14 days to assess changes in the joint diameter and to determine the VBS. (A) Knee joint diameter, before the injury and alternate days for 2 weeks following the injury, was measured using electronic calipers. The diameter of the knee joint before the injury was subtracted from the diameter following the injury, and the differences in the knee joint diameter were plotted as the percentage change in joint diameter. (B) Joint bleeding was evaluated by physical examination of knee joints and assigning an arbitrary score (0 to 3; 0, normal knee and absence of blood; 1, normal knee, and presence of blood; 2, distended but not a tense knee, and presence of blood; 3, tense and distended knee, and presence of blood). (C) Photographs of representative knee joints at the end of 14 days after the injury. Mice were euthanized, and the hind limbs were photographed after removing overlying skin. UN, an uninjured knee. (D) Synovitis score. Synovitis scores were assigned as described in “Materials and methods,” and the maximum synovitis score was 11. *Statistically significant difference in the data between mice administered with control IgG and mice administered MAPC 1591. #Statistically significant difference in the data between mice administered with MPC1609 and mice administered with MAPC1591. Data were plotted as ± standard error of the mean (SEM; n = 7 to 8 mice per group). *,#P < .05; **,##P < .01; ***P < .001; ****P < .0001.
Administration of MAPC1591, and not MPC1609, reduces joint bleeding, joint edema, and synovitis in hemophilia A mice following needle puncture joint injury. A single dose of control IgG, MPC1609, or MAPC1591 (1 mg/kg) was administered to FVIII−/− mice ip. Twenty-four hours later, joint bleeding was induced with a needle puncture. Mice were monitored for 14 days to assess changes in the joint diameter and to determine the VBS. (A) Knee joint diameter, before the injury and alternate days for 2 weeks following the injury, was measured using electronic calipers. The diameter of the knee joint before the injury was subtracted from the diameter following the injury, and the differences in the knee joint diameter were plotted as the percentage change in joint diameter. (B) Joint bleeding was evaluated by physical examination of knee joints and assigning an arbitrary score (0 to 3; 0, normal knee and absence of blood; 1, normal knee, and presence of blood; 2, distended but not a tense knee, and presence of blood; 3, tense and distended knee, and presence of blood). (C) Photographs of representative knee joints at the end of 14 days after the injury. Mice were euthanized, and the hind limbs were photographed after removing overlying skin. UN, an uninjured knee. (D) Synovitis score. Synovitis scores were assigned as described in “Materials and methods,” and the maximum synovitis score was 11. *Statistically significant difference in the data between mice administered with control IgG and mice administered MAPC 1591. #Statistically significant difference in the data between mice administered with MPC1609 and mice administered with MAPC1591. Data were plotted as ± standard error of the mean (SEM; n = 7 to 8 mice per group). *,#P < .05; **,##P < .01; ***P < .001; ****P < .0001.
We also assessed joint bleeding in mice for 14 days by visually inspecting mice on alternate days and assigning a VBS. No statistically significant differences were observed in the VBS among mice administered with control IgG, MPC1609, or MAPC1591 at day 2 (Figure 3B). However, the VBS has decreased steadily and significantly from day 3 onwards in FVIII−/− mice administered with MAPC1591, reaching close to zero by day 12, whereas the visual bleeding has remained persistent in FVIII−/− mice administered with control IgG or MPC1609 (Figure 3B). Gross examination of knee joints on day 14 showed a massive swelling of the joint observed at 5 hours after the injury (Figure 2A) was mostly resolved, but a significant amount of blood was still seen in the joint space of FVIII−/− mice administered with control IgG or MPC1609 (Figure 3C). The blood in the articular space is mostly resolved at day 14 in mice administered with MAPC1591 (Figure 3C).
Analysis of iron deposition in the synovium by Prussian blue staining of joint tissue sections as an indicator of recurrent joint bleeding showed prominent iron deposition in FVIII−/− mice treated with control IgG or MPC1609 (supplemental Figure 6A). Although initial bleeding was equal in all experimental treatments, minimal iron deposition was found in mice treated with MAPC1591 (supplemental Figure 6A). Quantitative determination of iron content in joint tissues using the Iron Assay Kit (Sigma) further confirmed that no iron was deposited in joints of hemophilia mice treated with MAPC1591 (supplemental Figure 6B). The global scoring of the synovitis pathology, based on the scoring system proposed by Valentino and colleagues,30 showed that the synovitis was less severe in mice treated with MAPC1591 compared with mice treated with control IgG or MPC1609 (Figure 3D).
Blocking APC anticoagulant activity without preventing APC cytoprotective function protects against HA following hemarthrosis
When knee joint tissue sections were analyzed histopathologically 14 days after the needle puncture, substantial synovial thickening with inflammation was observed in FVIII−/− mice treated with control IgG (Figure 4A-B). The synovium was infiltrated with macrophages (Figure 4C-D) and remodeled with neovascularization (Figure 4E-F). Administration of a single dose of MAPC1591 before the needle puncture–induced joint bleeding markedly reduced the synovial hyperplasia (Figure 4A-B), macrophage infiltration into the synovium (Figure 4C-D), and neovascularization (Figure 4E-F). However, administration of MPC1609 that inhibits both APC anticoagulant activity and cytoprotective signaling had no significant effect on the synovial hyperplasia, macrophage invasion, or neovascularization seen in joints following the needle puncture injury. Histopathological examination of other tissues showed that the administration of MPC1609 or MAPC1591 or the needle puncture injury of knees did not induce any pathological changes in other tissues (supplemental Figure 7). Analysis of joint tissue sections for cartilage degeneration by the Alcian blue staining revealed that administration of MAPC1591, but not MPC1609, prevented the degeneration of cartilage induced by joint bleeding (Figure 5A-B). Staining joint tissues with Safranin O/Fast Green, which also stains glycosaminoglycans and proteoglycans as with Alcian blue, further supported the observation that blocking APC anticoagulation activity with MAPC1591 reduces the cartilage destruction induced by a joint bleed (Figure 5C-D).
![Administration of MAPC1591 that selectively blocks anticoagulant activity and not signaling function of APC prevents joint bleed–induced hemophilic synovitis, inflammation, and neoangiogenesis. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg, ip). Twenty-four hours later, joint bleeding was induced with a needle puncture. Two weeks after the injury, mice were euthanized; knee joints were excised and fixed, and joint tissue sections were processed for histopathological analysis by staining with H&E (A). Two thin black arrows pointing to each other indicate the width of the synovial lining; the yellow arrowhead points out red blood cells, and the thick black arrows point out synovial villi. (B) The joint tissue pathology was quantified by scoring H&E-stained sections on a 0 to 6 scale for synovial hyperplasia (0, normal, <4 cell layers thick; 1, four to five layers thick; 2, six to seven layers thick; 3, more than seven layers thick; the presence of RBC [0, absent; 1, present], villus formation [0, absent; 1, present], and discoloration by hemosiderin [0, absent; 1, present]). (C) Immunostaining of joint tissue sections with macrophage marker F4/80. Arrows point out macrophages. (D) Macrophage infiltration was quantified on a 0 to 4 scale (0, absence of macrophages; 1, scattered macrophages; 2, line of macrophages; 3, a cluster of macrophages; 4, sheets of macrophages). (E) Immunostaining of tissue sections with CD31 antibody to identify neoangiogenesis. Arrows point out blood vessels. (F) The number of blood vessels counted per field. Top panel images were captured at ×4 magnification. The squared area was imaged at ×40 magnification (bottom panel). The images shown are representative, and the data in bar graphs are mean ± SEM (n = 7 to 8 mice per group). **P < .01; ***P < .001; ****P < .0001. Note: Data shown for no treatment control group (no tr.) in this figure, and other figures were from our earlier publication.8 They were shown to illustrate that control IgG treatment had no significant effect on joint bleed–induced pathology in FVIII−/− mice.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/18/10.1182_blood.2021013119/4/m_bloodbld2021013119f4.png?Expires=1770965282&Signature=SkIjqhWKes7DeUi9o-4eGWTL6ydMLbU41~kqOMcI1SKLo--5v-jy5Xvhzxcj9xVJMmZJaT-6twiiPBnMQI5VkXOssUfvLgjvlXVqBYm9mLn9zJUUS-LWjJsgije9cKqC7p7oS1PNPhlWfPaGp~z2ilHwvj2x-rU7S11LuBJZf5CmsSPrvNYwKKf2CFK-VzfcDRL6QDT8Gy4L6Lq9O3EvGwsxnh6JfIQ1dgwic3biVWN1WWrw4to2I-yPFVKw30uWmBzvJOWAFUDPWUtlVAz9g~EtNvP6rl7rmsGOcoHuxsdp3jj8-nHxMzgSj2o-OU0rEN1jI5GuC1r2qDe8DVDCbQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Administration of MAPC1591 that selectively blocks anticoagulant activity and not signaling function of APC prevents joint bleed–induced hemophilic synovitis, inflammation, and neoangiogenesis. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg, ip). Twenty-four hours later, joint bleeding was induced with a needle puncture. Two weeks after the injury, mice were euthanized; knee joints were excised and fixed, and joint tissue sections were processed for histopathological analysis by staining with H&E (A). Two thin black arrows pointing to each other indicate the width of the synovial lining; the yellow arrowhead points out red blood cells, and the thick black arrows point out synovial villi. (B) The joint tissue pathology was quantified by scoring H&E-stained sections on a 0 to 6 scale for synovial hyperplasia (0, normal, <4 cell layers thick; 1, four to five layers thick; 2, six to seven layers thick; 3, more than seven layers thick; the presence of RBC [0, absent; 1, present], villus formation [0, absent; 1, present], and discoloration by hemosiderin [0, absent; 1, present]). (C) Immunostaining of joint tissue sections with macrophage marker F4/80. Arrows point out macrophages. (D) Macrophage infiltration was quantified on a 0 to 4 scale (0, absence of macrophages; 1, scattered macrophages; 2, line of macrophages; 3, a cluster of macrophages; 4, sheets of macrophages). (E) Immunostaining of tissue sections with CD31 antibody to identify neoangiogenesis. Arrows point out blood vessels. (F) The number of blood vessels counted per field. Top panel images were captured at ×4 magnification. The squared area was imaged at ×40 magnification (bottom panel). The images shown are representative, and the data in bar graphs are mean ± SEM (n = 7 to 8 mice per group). **P < .01; ***P < .001; ****P < .0001. Note: Data shown for no treatment control group (no tr.) in this figure, and other figures were from our earlier publication.8 They were shown to illustrate that control IgG treatment had no significant effect on joint bleed–induced pathology in FVIII−/− mice.
Administration of MAPC1591 that selectively blocks anticoagulant activity and not signaling function of APC prevents joint bleed–induced hemophilic synovitis, inflammation, and neoangiogenesis. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg, ip). Twenty-four hours later, joint bleeding was induced with a needle puncture. Two weeks after the injury, mice were euthanized; knee joints were excised and fixed, and joint tissue sections were processed for histopathological analysis by staining with H&E (A). Two thin black arrows pointing to each other indicate the width of the synovial lining; the yellow arrowhead points out red blood cells, and the thick black arrows point out synovial villi. (B) The joint tissue pathology was quantified by scoring H&E-stained sections on a 0 to 6 scale for synovial hyperplasia (0, normal, <4 cell layers thick; 1, four to five layers thick; 2, six to seven layers thick; 3, more than seven layers thick; the presence of RBC [0, absent; 1, present], villus formation [0, absent; 1, present], and discoloration by hemosiderin [0, absent; 1, present]). (C) Immunostaining of joint tissue sections with macrophage marker F4/80. Arrows point out macrophages. (D) Macrophage infiltration was quantified on a 0 to 4 scale (0, absence of macrophages; 1, scattered macrophages; 2, line of macrophages; 3, a cluster of macrophages; 4, sheets of macrophages). (E) Immunostaining of tissue sections with CD31 antibody to identify neoangiogenesis. Arrows point out blood vessels. (F) The number of blood vessels counted per field. Top panel images were captured at ×4 magnification. The squared area was imaged at ×40 magnification (bottom panel). The images shown are representative, and the data in bar graphs are mean ± SEM (n = 7 to 8 mice per group). **P < .01; ***P < .001; ****P < .0001. Note: Data shown for no treatment control group (no tr.) in this figure, and other figures were from our earlier publication.8 They were shown to illustrate that control IgG treatment had no significant effect on joint bleed–induced pathology in FVIII−/− mice.
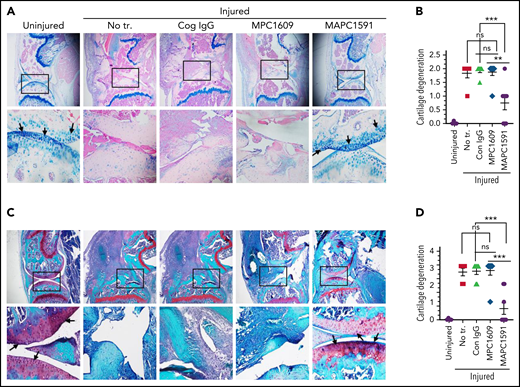
Treatment of FVIII−/− mice with an mAb that selectively inhibits APC anticoagulant activity (MAPC1591) reduces cartilage degeneration following a joint bleed. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg, ip). Twenty-four hours later, joint bleeding was induced with a needle puncture. Two weeks after the injury, mice were euthanized; knee joints were excised, and joint tissue sections were stained with Alcian blue (A) or Safranin O/Fast Green (C). Top panel images were captured at ×4 magnification. The squared area was imaged at ×20 magnification (bottom panel). Arrows point out the articular cartilage. Loss of blue stain (A) or red stain (B) in the articular cartilage region indicates the loss of glycosaminoglycans in the region and cartilage degeneration. (B) Cartilage degeneration in tissue sections stained with Alcian blue was scored on a 0 to 2 scale (0, absence of cartilage degeneration; 1, partial loss of proteoglycan content and pannus formation; 2, complete cartilage degeneration/absence of proteoglycans, pannus formation, and femur remodeling). (D) The quantified score of cartilage degeneration based on Safranin O/Fast Green Staining (0 to 3 scale; 0, normal/bright red staining of cartilage; 1, a slight reduction in staining; 2, a moderate reduction in staining; and 3, severe reduction/complete loss of staining). The data shown are mean ± SEM. ns, not statistically significant difference. **P < .01; ***P < 0 .001.
Treatment of FVIII−/− mice with an mAb that selectively inhibits APC anticoagulant activity (MAPC1591) reduces cartilage degeneration following a joint bleed. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg, ip). Twenty-four hours later, joint bleeding was induced with a needle puncture. Two weeks after the injury, mice were euthanized; knee joints were excised, and joint tissue sections were stained with Alcian blue (A) or Safranin O/Fast Green (C). Top panel images were captured at ×4 magnification. The squared area was imaged at ×20 magnification (bottom panel). Arrows point out the articular cartilage. Loss of blue stain (A) or red stain (B) in the articular cartilage region indicates the loss of glycosaminoglycans in the region and cartilage degeneration. (B) Cartilage degeneration in tissue sections stained with Alcian blue was scored on a 0 to 2 scale (0, absence of cartilage degeneration; 1, partial loss of proteoglycan content and pannus formation; 2, complete cartilage degeneration/absence of proteoglycans, pannus formation, and femur remodeling). (D) The quantified score of cartilage degeneration based on Safranin O/Fast Green Staining (0 to 3 scale; 0, normal/bright red staining of cartilage; 1, a slight reduction in staining; 2, a moderate reduction in staining; and 3, severe reduction/complete loss of staining). The data shown are mean ± SEM. ns, not statistically significant difference. **P < .01; ***P < 0 .001.
MAPC1591, but not MPC1609, treatment reduces joint bleed–induced chondrocyte apoptosis
TUNEL staining of joint tissue sections harvested at day 14 following needle puncture–induced joint bleed showed a substantial amount of chondrocyte apoptosis in the joint cartilage of FVIII−/− mice administered with control IgG (Figure 6A). Administration of MAPC1591 before joint bleed markedly reduced chondrocyte apoptosis (Figure 6A-B). In contrast, the administration of MPC1609 had no significant effect on joint bleed–induced chondrocyte apoptosis (Figure 6A-B).
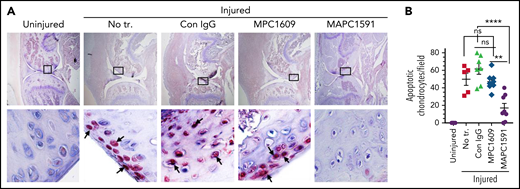
Inhibition of APC anticoagulant activity with MAPC1591 reduces joint bleed–induced chondrocyte apoptosis in hemophilia A mice. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg, ip). Twenty-four hours later, joint bleeding was induced by a needle puncture. Fourteen days following the injury, the knee joint was excised, sectioned, and stained with TUNEL. Red staining of chondrocytes, pointed out by arrows, indicates apoptotic chondrocytes. Top panel images were captured at ×4 magnification. The squared area was imaged at ×60 magnification (bottom panel). (B) The number of apoptotic chondrocytes was counted per high magnification field and quantified. The data shown are ± SEM. ns, not statistically significant difference. **P < .01; ****P < .0001.
Inhibition of APC anticoagulant activity with MAPC1591 reduces joint bleed–induced chondrocyte apoptosis in hemophilia A mice. FVIII−/− mice were administered with control IgG, MPC1609, or MAPC1591 (1 mg/kg, ip). Twenty-four hours later, joint bleeding was induced by a needle puncture. Fourteen days following the injury, the knee joint was excised, sectioned, and stained with TUNEL. Red staining of chondrocytes, pointed out by arrows, indicates apoptotic chondrocytes. Top panel images were captured at ×4 magnification. The squared area was imaged at ×60 magnification (bottom panel). (B) The number of apoptotic chondrocytes was counted per high magnification field and quantified. The data shown are ± SEM. ns, not statistically significant difference. **P < .01; ****P < .0001.
Treatment of FVIII−/− mice with MAPC1591, but not MPC1609, blocks joint bleed–induced elevated IL-6 expression and vascular leakage in the synovium
Hemarthrosis led to a marked increase in IL-6 levels in the synovium of FVIII−/− mice (Figure 7A). Treatment of FVIII−/− mice with MPC1609 or control IgG before the induction of joint bleeding with needle puncture did not affect IL-6 expression in the synovial fluid (Figure 7A). In contrast, the administration of MAPC1591 completely prevented IL-6 expression in the synovium following hemarthrosis (Figure 7A). In additional studies, we assessed barrier permeability in the injured knee at 14 days following needle puncture injury. The injury increased vascular leakage into knee joints and the surrounding tissue, and treatment of mice with MAPC1591, but not MPC1609, significantly reduced the vascular leakage (Figure 7B-C). Finally, fewer deaths were observed in FVIII−/− mice treated with MAPC1591 than FVIII−/− mice treated with control IgG or MPC1609 following the needle puncture–induced joint bleeding (supplemental Figure 8).
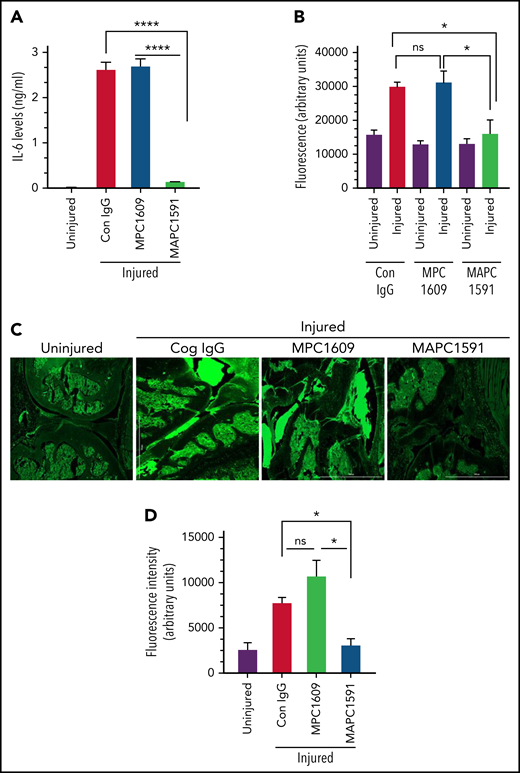
Selective blocking of APC anticoagulant activity prevents the elaboration of IL-6 and the vascular leakage in the synovium of FVIII−/− mice following the knee injury. FVIII−/− mice were administered with a control IgG, MAPC1591 that blocks the anticoagulant activity of APC, or MPC1609 that blocks both the anticoagulant and the signaling functions of APC (1 mg/kg, ip). After 24 hours, joint bleeding was induced by a needle puncture injury. (A) Seven days after the injury, synovial fluids were collected, and IL-6 levels in the synovial fluids were determined by ELISA. (n = 4 mice per group). (B-C) Fourteen days after the injury, vascular leakage was evaluated by extravasation of IV injected fluorescein dextran into knee joints. Fourteen days after the injury, mice were injected with fluorescein dextran (70 000 molecular weight, 20 mg/kg) IV via the tail vein. Mice were euthanized 3 hours following dextran administration and perfused with ice-cold saline supplemented with 5 mM CaCl2 and 1 mM MgCl2. Joint tissue was excised, and fluorescence intensities of joint tissue extracts were measured (corrected for autofluorescence) (B). Joint tissue sections were also analyzed by fluorescence microscopy at ×4 magnification (C), and the fluorescence intensity of tissue sections was quantified and corrected to autofluorescence (D). *P < .05; ****P < .0001; no statistically significant difference (n = 4).
Selective blocking of APC anticoagulant activity prevents the elaboration of IL-6 and the vascular leakage in the synovium of FVIII−/− mice following the knee injury. FVIII−/− mice were administered with a control IgG, MAPC1591 that blocks the anticoagulant activity of APC, or MPC1609 that blocks both the anticoagulant and the signaling functions of APC (1 mg/kg, ip). After 24 hours, joint bleeding was induced by a needle puncture injury. (A) Seven days after the injury, synovial fluids were collected, and IL-6 levels in the synovial fluids were determined by ELISA. (n = 4 mice per group). (B-C) Fourteen days after the injury, vascular leakage was evaluated by extravasation of IV injected fluorescein dextran into knee joints. Fourteen days after the injury, mice were injected with fluorescein dextran (70 000 molecular weight, 20 mg/kg) IV via the tail vein. Mice were euthanized 3 hours following dextran administration and perfused with ice-cold saline supplemented with 5 mM CaCl2 and 1 mM MgCl2. Joint tissue was excised, and fluorescence intensities of joint tissue extracts were measured (corrected for autofluorescence) (B). Joint tissue sections were also analyzed by fluorescence microscopy at ×4 magnification (C), and the fluorescence intensity of tissue sections was quantified and corrected to autofluorescence (D). *P < .05; ****P < .0001; no statistically significant difference (n = 4).
Discussion
Clinical trials and studies in animal model systems in the last few years suggest that blocking natural anticoagulants is a promising strategy to treat patients with hemophilia with and without inhibitors.14,16,18,22,24 Recent studies provided a proof of concept that inhibition of endogenous APC anticoagulant activity corrects bleeding in hemophilia animal model systems.18,24 However, these studies were primarily limited to acute bleeding models. We are not aware of any studies that examine whether inhibition of endogenous APC activity prevents recurrent joint bleeding and the development of HA. Here, we provide convincing evidence that selective inhibition of endogenous APC anticoagulant activity with an mAb provides robust protection against the development of HA following the needle puncture–induced joint bleeding. Interestingly, inhibition of both the anticoagulant activity and cytoprotective functions of APC failed to protect against HA. These data suggest that inhibition of the APC anticoagulant pathway is an effective strategy in preventing HA in patients with hemophilia, but the preservation of endogenous APC’s cytoprotective signaling is essential for this strategy.
MAPC1591 and MPC1609 were originally developed to investigate the relative importance of anticoagulant and anti-inflammatory effects of endogenous APC in mice.28 MPC1609 binds to protein C and APC and inhibits their binding to the endothelium. MPC1609 inhibits both anticoagulant and cytoprotective activities of APC, probably binding to the Gla domain of protein C/APC, a region known to be necessary for binding to both EPCR and phospholipids.44 MAPC1591 specifically binds APC without interfering with its binding to the endothelium. It inhibits APC anticoagulant activity, presumably by interfering with APC binding to its substrate FVa. MAPC1591 does not inhibit APC’s signaling function.28 It was found that administration of MPC1609 exacerbates a sublethal dose of LPS into lethality, whereas MAPC1591 protects against LPS-induced sepsis and lethality.28 Similar results were obtained with these antibodies in gram-negative pneumosepsis45 and renal ischemia/reperfusion injury.46 In our studies, although administration of MPC1609 failed to prevent HA, we found no adverse effects of the antibody. The antibody exacerbated neither joint inflammation nor the severity of HA. Differences in inflammatory injuries may be responsible for the above differences.
The marked differences between MAPC1591 and MPC1609 mAb in preventing HA are not due to potential differences between their anticoagulant activities, as they are equally effective in correcting acute bleeding induced by the saphenous vein incision. Measurement of the antibody levels in plasma after 7 days showed no significant differences between the levels of MPC1609 (138 ± 50 ng/mL) and MAPC1591 (132 ± 11 ng/mL), ruling out that differences in their pharmacokinetics are responsible for the observed differences. These data suggest that preserving APC’s cytoprotective signaling function while inhibiting APC anticoagulant activity is essential in preventing HA. At present, it is difficult to precisely evaluate the relative contribution of APC cytoprotective effect and the enhanced coagulation activity from the inhibition of APC anticoagulant activity in suppressing HA in FVIII−/− mice treated with MAPC1591. The lack of antibodies that selectively inhibit APC’s signaling without interfering with APC’s anticoagulant activity makes such investigation unfeasible currently. The observation that inhibition of APC anticoagulant activity restores hemostasis and prevents HA in FVIII−/− mice suggests that pathophysiologic relevant amounts of APC were generated in hemophilia despite hemophilic condition severely impairs thrombin generation, an enzyme that activates protein C.47 It is possible that inhibition of endogenous APC’s anticoagulant activity that increases thrombin generation might, in turn, lead to increased generation of APC, particularly at the injury site, which is sufficient to exert effective cryoprotection. Currently, it is difficult to test this possibility as limitations in accurately measuring sub-nanograms of APC and minute amounts of synovial fluid in the murine model.
The mechanism by which APC-mediated cytoprotective signaling protects against HA is unclear. The pathogenesis of HA is unique and complex.4,6,7,9 It is influenced by the repeated release of hemoglobin and iron deposition in the joint from recurrent bleeding. Accumulation of iron in the joint triggers synovial inflammation and induces hyperreactive changes in the synovium, including hypertrophy and villi formation. The inflamed and hypertrophic synovium induces the release of growth factors, such as VEGF, that promote increased vascular leakage and neoangiogenesis. Synovial derived proinflammatory cytokines and matrix metalloproteinases combined with extracellular matrix degradation and chondrocyte apoptosis induced by direct exposure to blood cause cartilage degeneration. The progression of synovitis and cartilage degeneration ultimately leads to bone damage. APC cytoprotective signaling could affect the pathogenesis of HA at multiple levels. APC-induced cytoprotective signaling was shown to suppress inflammation, stabilize vascular barrier integrity, and inhibit apoptosis.36 The barrier-protective effect of APC may stabilize the fragile barrier integrity of the synovium in hemophilia and thus prevent recurrent bleeding into joints. Attenuation of rebleeding into joints would block the vicious cycle of accumulation of iron in joints, inflammation, hypertrophy, and neoangiogenesis and thus prevent the development of HA. The data presented in the paper, which show reduced vascular leakage and no significant deposition of iron in the synovium in mice administered with MAPC1591, support the above concept.
Current data on the importance of preserving endogenous APC signaling, in addition to inhibiting APC anticoagulant pathway, in suppressing HA differs on the face value from our earlier data obtained using EPCR blocking mAb.8 The earlier study showed convincingly that administration of EPCR blocking mAb attenuated HA in FVIII−/− mice. EPCR blocking mAb prevents both protein C and APC binding to EPCR and thus blocks APC anticoagulant pathway by curtailing APC generation as well as EPCR-dependent APC signaling.25,26,48 Therefore, data from our earlier study suggest that preservation of EPCR-APC signaling is not essential for suppressing HA. Reconciliation of contrasting data of the current and earlier study raises the possibility that low, but pathophysiologic relevant amounts of APC may be generated independent of EPCR, and EPCR-independent APC signaling might contribute to cytoprotective functions of APC that suppress HA. Alternatively, it is possible that downregulation of APC generation in EPCR-deficient mice could enhance the coagulation to such an extent that it would prevent chronic and recurrent bleeding into joints and thus the development of HA. Under this scenario, the cytoprotective signaling functions of EPCR-APC could be redundant.
Regarding the possibility of generating APC and exerting the cytoprotective effects independent of EPCR, it may be pertinent to point out that integrin-β3 in podocytes was shown to substitute for known functions of EPCR on endothelial cells, that is, protein C activation and APC signaling.38,49 Several other studies showed that APC could also mediate cytoprotective signaling in certain cell types partially or fully independent of EPCR.38,41,42,50-52 At present, it is unclear whether APC’s cytoprotective signaling in the synovium is dependent on EPCR or other APC’s putative receptors, such as ApoER2, Mac-1, or integrin-β3.53 EPCR and PAR1 expression was found in many cell types associated with the synovium, including cells in the synovial lining layer, synovial fibroblasts, monocytes/macrophages, and chondrocytes.32,54-58 However, little is known about the expression of putative APC receptors in these cell types and their potential role in mediating APC cytoprotective signaling. Detailed studies are needed to characterize the expression of EPCR, PARs, and various APC’s putative receptors in various synovial cell types and their signaling capabilities to ascertain exact mechanisms by which APC protects HA. Such studies are beyond the scope of the present investigation.
In summary, our current data provide convincing evidence that selective inhibition of APC anticoagulant function by an inhibitory mAb is highly effective in preventing HA in a mouse model. These data suggest that specific inhibition of APC’s anticoagulant activity could be an effective and viable strategy to treat patients with hemophilia to prevent recurrent joint bleeding and protect against HA. The possibility that such a treatment could be used to treat all categories of hemophilia makes it an attractive option.
Acknowledgments
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL107483 and HL124055 (L.V.M.R.) and a Judith Graham Pool's postdoctoral fellowship from the National Hemophilia Foundation (J.M., K.D.).
Authorship
Contribution: J.M. performed most of the experiments, analyzed data, and prepared an initial draft of the manuscript; S.K. performed saphenous vein incision-induced bleeding experiments; V.K. and K.D. performed in vitro experiments related to the characterization of the antibodies; C.T.E. provided critical reagents to the study; U.R.P. contributed to the study design, data analysis, and manuscript preparation; L.V.M.R. conceived and designed the research, analyzed data, and wrote the manuscript; and all authors contributed to the preparation of the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: L. Vijaya Mohan Rao, Department of Cellular and Molecular Biology, Center for Biomedical Research, The University of Texas Health Science Center at Tyler, 11937 US Highway 271, Tyler, TX 75708-3154; e-mail: vijay.rao@uthct.edu.
Requests for data sharing may be submitted to L. Vijaya Mohan (vijay.rao@uthct.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal