


Abstract
The standard of care treatment strategy for patients with relapsed or refractory large B-cell lymphoma (LBCL) has been high-dose chemotherapy followed by autologous stem cell transplantation (ASCT) if chemotherapy sensitive in suitable patients. Because of treatment intensity, this approach has only been feasible in half of patients and because of chemotherapy resistance has only been successful in a quarter of transplant-eligible patients. Chimeric antigen receptor (CAR) T-cell therapy, using genetically modified autologous T cells targeting CD19, has been approved for third-line therapy of LBCL and has been associated with durable remissions in a proportion of patients. In this review, we interpret the design and results of 3 randomized phase 3 trials comparing CAR T-cell therapy and ASCT and their implications for CAR T-cell therapy as a potential new standard of care for second-line treatment in appropriate patients with refractory or early relapsing LBCL.
Introduction
It is all too common for new advances to be described as a paradigm shift. The word paradigm is defined as a model or pattern, but the root is from the Greek words para and deiknynai, translated as to “show side by side.” In this review, we will show side by side how the paradigm of chemotherapy and autologous stem cell transplantation (ASCT) may shift to chimeric antigen receptor (CAR) T-cell therapy in some patients with large B-cell lymphoma (LBCL) receiving second-line therapy based on 3 recently completed randomized phase 3 trials.1-3
LBCL is a heterogenous category of clinicopathologic entities exhibiting aggressive behavior, of which diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS) is the most common.4 The most recent iteration of the World Health Organization classification now recognizes a separate category of high-grade B-cell lymphoma (with MYC and BCL2 and/or BCL6 rearrangements or NOS) that for the purposes of this article will be included under the heading of LBCL. Although most patients with LBCL can be cured with immunochemotherapy, nearly 40% will be refractory to or relapse (R/R) after initial treatment.5 Most patients experience treatment failure within the first 2 years, with outcomes correlated with timing of progression or relapse.6 Although patients can be cured with secondary therapies, most patients die from their disease. Approximately 10% to 15% of patients exhibit primary refractory disease, typically defined as an incomplete response or relapse within 3 to 6 months. With inherent chemotherapy resistance, these patients have the poorest outcomes, with median overall survival (OS) of about 6 months.7 Patients with late relapses beyond 2 years can have more favorable outcomes and may have biologically distinct disease.8,9
Standard of care therapy for R/R LBCL
For a quarter century, the standard treatment strategy for patients with R/R LBCL has been to attempt high-dose chemotherapy and ASCT, based on the premise that treatment resistance may be overcome with higher doses of cytotoxic therapy. Proceeding to ASCT with chemotherapy-insensitive disease was shown to be futile; hence, the requirement was added to demonstrate a response to salvage therapy before proceeding to transplant.10 The Parma trial randomized chemotherapy-sensitive patients with R/R intermediate to high-grade non-Hodgkin lymphoma to ASCT or DHAP (dexamethasone, high-dose cytarabine, and cisplatin).11 ASCT induced greater toxicity, but improved efficacy, with event-free survival (EFS) at 5 years of 46% in comparison with 12% with DHAP. Despite occurring before the introduction of rituximab, this trial established ASCT as standard of care, which has prevailed until today.
The strategy of ASCT has numerous limitations. Given the intensity, only half of R/R patients are transplant eligible because of advanced age or comorbidities. An additional half of transplant-eligible patients will prove insensitive to salvage therapy, precluding ASCT. The most commonly used platinum-based salvage regimens, rituximab (R)-DHAP, R-ICE (ifosfamide, carboplatin, and etoposide), and R-GDP (gemcitabine, cisplatin, dexamethasone) have shown similar efficacy, with response rates in the range of 44% to 63%.12,13 The CORAL trial, comparing R-DHAP and R-ICE, identified a poor-risk population with prior exposure to rituximab and R/R disease within 12 months of diagnosis, with 3-year progression-free survival (PFS) of 23%.12 The LY.12 trial, comparing DHAP and GDP, similarly demonstrated inferior outcomes in patients with primary refractory disease or an initial response duration of less than 12 months.13 Thus, in the rituximab era, the benefit of an ASCT approach may be diminished, particularly for patients with refractory or early relapsing disease, which represents the majority of patients. Perhaps the biggest limitation of this approach is that it relies on cytotoxic chemotherapy in the setting of proven resistance. Resistance mechanisms are likely multifactorial, including biological, immunologic, and host factors.14 Novel therapies have demonstrated the ability to overcome chemotherapy resistance in transplant-ineligible patients with R/R DLBCL, and thus it is logical to explore whether these may improve on ASCT, particularly in high-risk populations.15-21
CAR T-cell therapy
CAR T-cell therapy is a genetically modified cellular treatment that was named the “2018 Advance of the Year” by the American Society of Clinical Oncology.22 The initial products approved, axicabtagene ciloleucel (axi-cel), tisagenlecleucel (tisa-cel), and lisocabtagene maraleucel (liso-cel) involve autologous T cells programmed to express a CAR targeting the B-cell marker CD19.15-17 These products have design differences, including differences in the costimulatory domain, mechanism of viral transfection, ability for cryopreservation, and need for T-cell selection (Figure 1). Updated results from the single arm phase 2 pivotal trials, ZUMA-1, Juliet, and Transform, that established their utility in patients with multiply relapsed LBCL are summarized in Figure 1. All 3 CAR T-cell products demonstrated capacity to induce durable remissions in approximately one-third of treated patients (including patients who had not had a durable remission with a prior ASCT) and have been US Food and Drug Administration approved for patients with R/R LBCL after at least 2 lines of therapy. Registry data of patients treated in clinical care has confirmed this benefit.15-17,23-36 As these products have never been compared head-to-head, their comparative effectiveness has not been determined.

Distinguishing features of the US Food and Drug Administration–approved CAR T-cell constructs and updated outcomes from the pivotal trials.25,26,28,36
Distinguishing features of the US Food and Drug Administration–approved CAR T-cell constructs and updated outcomes from the pivotal trials.25,26,28,36
CAR T-cell therapy provides a new option with potential to achieve durable remission and possibly cure in patients with multiply relapsed LBCL, where this was previously unlikely. However, CAR T-cell therapy is associated with new logistical challenges and unique toxicities. It is a multistep process that requires careful coordination of care between primary oncologists and treating centers. Patients must undergo leukapheresis and then wait several weeks for product generation before receiving lymphodepleting therapy (most commonly cyclophosphamide and fludarabine) and CAR T-cell infusion. Patients with symptomatic or rapidly progressing disease may require temporizing treatment, commonly referred to as bridging therapy, to control disease until the CAR T-cell product is available for infusion. Although similar regimens may be used, the goal of bridging therapy is different than salvage chemotherapy before ASCT, in that it is primarily used to alleviate symptoms and minimize disease progression to maintain CAR T-cell eligibility. The use of bridging therapy has been associated with poorer outcomes, but this is likely a surrogate for more aggressive or higher burden disease.37,38 CAR T-cell toxicities of concern include cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicty syndrome (ICANS).39 CRS is driven by a supraphysiologic cytokine elevation associated with CAR T-cell expansion soon after infusion and can cause fever, hypotension, and risk of multiorgan failure. The interleukin-6 inhibitor tociluzimab can ameliorate CRS and is typically used when grade 2 or higher symptoms develop.39,40 ICANS is a unique toxicity presenting with a spectrum of manifestations including delirium, encephalopathy, aphasia, and occasionally seizures. Management of ICANS is largely supportive, with symptoms being transient in most cases. Toxicities may vary across the available products, but direct comparison is difficult because of differences between patient populations and grading scales used. Rates of CRS and ICANS observed within the pivotal trials are outlined in Figure 1.15-17 Additional toxicities of concern include infection, hypogammaglobulinemia, and prolonged cytopenias.41
Rationale for and design of phase 3 CAR T-cell vs ASCT trials
Given the promising results within phase 2 pivotal trials and favorable comparisons with standard of care outcomes in the CORAL and LY.12 trials as a benchmark, it was hypothesized that CAR T-cell therapy may achieve superior outcomes in comparison with the treatment strategy of salvage therapy followed by ASCT as second-line therapy in patients with high-risk R/R LBCL. Three randomized phase 3 trials were launched to evaluate this hypothesis: ZUMA-7 (axi-cel, NCT03391466), Belinda (tisa-cel, NCT03570892), and Transform (liso-cel, NCT03575351).1-3
The 3 trials have numerous similarities, including enrolling adult patients with LBCL refractory to or relapsed within 12 months from completion of first-line therapy, who were considered candidates for ASCT by the treating physician. Key trial design features are described in Table 1. Main eligibility criteria were similar, with the exception of an upper age limit of 75 years on Transform. Belinda and Transform performed leukapheresis on all patients before randomization to allow for crossover from standard of care, whereas crossover was not planned per protocol on ZUMA-7, but patients could potentially access CAR T-cell therapy off-protocol. All trials allowed a choice of salvage regimens including R-DHAP, R-ICE, and R-GDP. Belinda and Transform allowed bridging chemotherapy before CAR T-cell therapy with the same regimens from the standard of care arm. However, Transform allowed only 1 cycle of bridging, whereas Belinda allowed more than 1 cycle, as well as switching to a second regimen. Importantly, ZUMA-7 did not allow bridging chemotherapy but permitted corticosteroids for disease stabilization.
Trial design features
| ZUMA-7 | Belinda | Transform | |
|---|---|---|---|
| Histologies included | DLBCL NOS,* including transformed from FL, HGBCL with or without MYC and BCL2/6, T/H-RLBCL, Primary cutaneous DLBCL - leg type | DLBCL NOS, including transformed from indolent NHL, HGBCL with or without MYC and BCL2/6, T/H-RLBCL, Primary cutaneous DLBCL - leg type FL grade 3B, PMBCL, Intravascular LBCL, ALK + LBCL, HHV8 + LBCL | DLBCL NOS, including transformed from indolent NHL, HGBCL with MYC and BCL2/6, T/H-RLBCL, FL grade 3B, PMBCL |
| Product | Axi-cel, CD28/CD3zeta 2 × 106 cells/kg | Tisa-cel, 4 – 1BB/CD3zeta 0.6-6 × 108 cells | Liso-cel, 4 − 1BB/CD3zeta 1 × 108 cells |
| 1L refractory definition | • PD as best response • SD after at least 4 cycles • PR with + biopsy or PD <12 mo from 1L start | • PD/SD as best response | • PD/SD/PR as best response • CR with progression <3 mo |
| 1L relapsed definition | • CR followed by + biopsy <12 mo from 1L end | • Positive biopsy ≤12 mo from 1L end | • CR followed by + biopsy 3-12 mo from 1L end |
| Age | 18+ | 18+ | 18-75 |
| Leukapheresis time point | • At randomization • Only CAR T-cell arm | • Before randomization • All patients | • Before randomization • All patients |
| Stratification factors | 1. Refractory vs Relapse ≤6 mo vs Relapse >6-12 mo 2. 2L AAIPI 0-1 vs 2-3 | 1. Refractory or relapsed ≤6 mo vs relapsed 6-12 mo 2. IPI <2 vs ≥2 | 1. Refractory vs relapse 2. 2L AAIPI 0-1 vs 2-3 |
| Bridging therapy | • Dexamethasone ≤40 mg for ≤4 d | • R-ICE • R-GDP • R-DHAP • R-GemOx | • R-ICE • R-GDP • R-DHAP |
| LD chemotherapy | • Fludarabine 30 mg/m2 × 3 d • Cyclophosphamide 500 mg/m2 × 3 d | • Fludarabine 25 mg/m2 × 3 d and • Cyclophosphamide 250 mg/m2 × 3d OR • Bendamustine 90 mg/m2 × 2 d | • Fludarabine 30 mg/m2 × 3 d • Cyclophosphamide 300 mg/m2 × 3 d |
| SOC chemotherapy | • R-ICE • R-GDP • R-DHAP • R-ESHAP | • R-ICE • R-GDP • R-DHAP • R-GemOx | • R-ICE • R-GDP • R-DHAP |
| Crossover to CAR T-cell therapy | No | Yes, if • <PR/CR by 12 wk (after 2 SOC regimens) • PD at any time | Yes, if • <PR/CR by 9 wk • PD at any time • Need for new therapy after 18 wk |
| EFS definition | Time from randomization to: • PD • Death • <PR at day 150 assessment • Start of new lymphoma therapy | Time from randomization to: • PD • Death • <PR at/after week 12 | Time from randomization to: • PD • Death • ≤PR by week 9 • Start of new lymphoma therapy |
| ZUMA-7 | Belinda | Transform | |
|---|---|---|---|
| Histologies included | DLBCL NOS,* including transformed from FL, HGBCL with or without MYC and BCL2/6, T/H-RLBCL, Primary cutaneous DLBCL - leg type | DLBCL NOS, including transformed from indolent NHL, HGBCL with or without MYC and BCL2/6, T/H-RLBCL, Primary cutaneous DLBCL - leg type FL grade 3B, PMBCL, Intravascular LBCL, ALK + LBCL, HHV8 + LBCL | DLBCL NOS, including transformed from indolent NHL, HGBCL with MYC and BCL2/6, T/H-RLBCL, FL grade 3B, PMBCL |
| Product | Axi-cel, CD28/CD3zeta 2 × 106 cells/kg | Tisa-cel, 4 – 1BB/CD3zeta 0.6-6 × 108 cells | Liso-cel, 4 − 1BB/CD3zeta 1 × 108 cells |
| 1L refractory definition | • PD as best response • SD after at least 4 cycles • PR with + biopsy or PD <12 mo from 1L start | • PD/SD as best response | • PD/SD/PR as best response • CR with progression <3 mo |
| 1L relapsed definition | • CR followed by + biopsy <12 mo from 1L end | • Positive biopsy ≤12 mo from 1L end | • CR followed by + biopsy 3-12 mo from 1L end |
| Age | 18+ | 18+ | 18-75 |
| Leukapheresis time point | • At randomization • Only CAR T-cell arm | • Before randomization • All patients | • Before randomization • All patients |
| Stratification factors | 1. Refractory vs Relapse ≤6 mo vs Relapse >6-12 mo 2. 2L AAIPI 0-1 vs 2-3 | 1. Refractory or relapsed ≤6 mo vs relapsed 6-12 mo 2. IPI <2 vs ≥2 | 1. Refractory vs relapse 2. 2L AAIPI 0-1 vs 2-3 |
| Bridging therapy | • Dexamethasone ≤40 mg for ≤4 d | • R-ICE • R-GDP • R-DHAP • R-GemOx | • R-ICE • R-GDP • R-DHAP |
| LD chemotherapy | • Fludarabine 30 mg/m2 × 3 d • Cyclophosphamide 500 mg/m2 × 3 d | • Fludarabine 25 mg/m2 × 3 d and • Cyclophosphamide 250 mg/m2 × 3d OR • Bendamustine 90 mg/m2 × 2 d | • Fludarabine 30 mg/m2 × 3 d • Cyclophosphamide 300 mg/m2 × 3 d |
| SOC chemotherapy | • R-ICE • R-GDP • R-DHAP • R-ESHAP | • R-ICE • R-GDP • R-DHAP • R-GemOx | • R-ICE • R-GDP • R-DHAP |
| Crossover to CAR T-cell therapy | No | Yes, if • <PR/CR by 12 wk (after 2 SOC regimens) • PD at any time | Yes, if • <PR/CR by 9 wk • PD at any time • Need for new therapy after 18 wk |
| EFS definition | Time from randomization to: • PD • Death • <PR at day 150 assessment • Start of new lymphoma therapy | Time from randomization to: • PD • Death • <PR at/after week 12 | Time from randomization to: • PD • Death • ≤PR by week 9 • Start of new lymphoma therapy |
1L, first-line; 2L, second-line; AAIPI, age-adjusted international prognostic index; ALK, anaplastic lymphoma kinase; BCL2/6, B-cell lypmphoma protein 2 and/or 6; FL, follicular lymphoma; HGBCL, high-grade B-cell lymphoma; HHV8, human herpesvirus-8; IPI, international prognostic index; LD, lymphodepleting chemotherapy; PD, progressive disease; PMBCL, primary mediastinal B-cell lymphoma; PR, partial response; R-ESHAP, rituximab, etoposide, steroids, high-dose cytarabine, cisplatin; R-GemOx, rituximab, gemcitabine, oxaliplatin; SD, stable disease; SOC, standard of care; T/H-RLBCL, T-cell/histiocyte-rich large B-cell lymphoma.
The ZUMA7 protocol does not specifically state LBCL subtypes, but the subtypes listed are included in patients enrolled.
The primary end point for all 3 trials was EFS, although the definition differed. In addition to death and disease progression, all 3 trials included stable disease at different time points as events (ZUMA-7: day 150, Belinda: week 12, Transform: week 9). Although initiation of new antilymphoma therapy was considered an event within ZUMA-7 and Transform, Belinda did not include initiation of a second salvage or bridging regimen within the first 12 weeks as an event.
Patients and results
The 3 trials enrolled patients internationally at centers with expertise in both CAR T-cell therapy and ASCT. Patient characteristics are summarized in Table 2, and outcomes are described in Table 3.
Patient characteristics
| ZUMA-7 | Belinda | Transform | ||||
|---|---|---|---|---|---|---|
| Axi-cel | SOC | Tisa-cel | SOC | Liso-cel | SOC | |
| N | 180 | 179 | 162 | 160 | 92 | 92 |
| Median age, y (range) | 58 (21-80) | 60 (26-81) | 60 (19-79) | 58 (19-77) | 60 (20-74) | 58 (26-75) |
| Age ≥65 y, n (%) | 51 (28) | 58 (32) | 54 (33) | 46 (29) | 36 (39) | 25 (27) |
| Female sex (%) | 70 (39) | 52 (29) | 59 (36) | 62 (39) | 48 (52) | 31 (34) |
| AAIPI* 2-3IPI ≥2 (%) | 82 (46)* | 79 (44)* | 106 (65) | 92 (58) | 36 (39)* | 37 (40)* |
| GCB subgroup (%) | 109 (61) | 99 (55) | 46 (28) | 63 (39) | NR | NR |
| ABC subgroup (%) | 16 (9) | 9 (5) | 52 (32) | 42 (26) | NR | NR |
| HGBCL (%) | 31 (17) | 25 (14) | 32 (20) | 19 (12) | 22 (24) | 21 (23) |
| Refractory to 1L (%) | 133 (74) | 131 (73) | 107 (66) | 107 (67) | 67 (73) | 68 (74) |
| ZUMA-7 | Belinda | Transform | ||||
|---|---|---|---|---|---|---|
| Axi-cel | SOC | Tisa-cel | SOC | Liso-cel | SOC | |
| N | 180 | 179 | 162 | 160 | 92 | 92 |
| Median age, y (range) | 58 (21-80) | 60 (26-81) | 60 (19-79) | 58 (19-77) | 60 (20-74) | 58 (26-75) |
| Age ≥65 y, n (%) | 51 (28) | 58 (32) | 54 (33) | 46 (29) | 36 (39) | 25 (27) |
| Female sex (%) | 70 (39) | 52 (29) | 59 (36) | 62 (39) | 48 (52) | 31 (34) |
| AAIPI* 2-3IPI ≥2 (%) | 82 (46)* | 79 (44)* | 106 (65) | 92 (58) | 36 (39)* | 37 (40)* |
| GCB subgroup (%) | 109 (61) | 99 (55) | 46 (28) | 63 (39) | NR | NR |
| ABC subgroup (%) | 16 (9) | 9 (5) | 52 (32) | 42 (26) | NR | NR |
| HGBCL (%) | 31 (17) | 25 (14) | 32 (20) | 19 (12) | 22 (24) | 21 (23) |
| Refractory to 1L (%) | 133 (74) | 131 (73) | 107 (66) | 107 (67) | 67 (73) | 68 (74) |
ABC, activated B-cell; GCB, germinal center B cell; HGBCL, high grade B-cell lymphoma; NR, not reported; SOC, standard of care.
Age-adjusted international prognostic index.
Outcomes
| ZUMA-7 | Belinda | Transform | ||||
|---|---|---|---|---|---|---|
| Axi-Cel | SOC | Tisa-Cel | SOC | Liso-Cel | SOC | |
| Received bridging corticosteroids (%) | 36 | — | — | — | — | — |
| Received bridging chemotherapy (%) | 0 | — | 83 | — | 63 | — |
| Received >1 SOC chemotherapy regimen (%) | — | 0 | — | 54 | — | 0 |
| Received intended CAR T cell (%) | 94 | — | 96 | — | 97.8 | — |
| Median time to CAR T-cell infusion in days, (interquartile range* or range†) | 29 (27-34)* | — | 52 (31-135)† | — | NR | — |
| Received intended ASCT (%) | — | 36 | — | 32.5 | — | 45.6 |
| Crossover pn protocol (%) | — | — | — | 51 | — | 51 |
| Received cellular therapy off protocol (%) | — | 56 | — | — | — | |
| Follow up, median in months | 24.9 | 10 | 6.2 | |||
| ORR/CR rate (%) | 83/65 | 50/32 | 46/28 | 43 /28 | 86/66 | 48/39 |
| EFS, median in months | 8.3 | 2 | 3 | 3 | 10.1 | 2.3 |
| EFS, % (timepoint in months) | 41 (24 mo) | 16 (24 mo) | NR | NR | 63 (6 mo) | 33 (6 mo) |
| EFS HR (95% CI) | 0.4 (0.31-0.51) | 1.07 (0.82-1.4) | 0.35 (0.23-0.53) | |||
| PFS, median in months | 14.7 | 3.7 | NR | NR | 14.8 | 5.7 |
| PFS HR (95% CI) | 0.49 (0.37-0.65) | NR | 0.406 (0.21-0.66) | |||
| OS, median in months | NE | 25.7 | 16.9 | 15.3 | NE | 16.4 |
| OS HR (95% CI) | 0.708 (0.515-0.972)‡ | NR | 0.51 (0.26-1.004) | |||
| ZUMA-7 | Belinda | Transform | ||||
|---|---|---|---|---|---|---|
| Axi-Cel | SOC | Tisa-Cel | SOC | Liso-Cel | SOC | |
| Received bridging corticosteroids (%) | 36 | — | — | — | — | — |
| Received bridging chemotherapy (%) | 0 | — | 83 | — | 63 | — |
| Received >1 SOC chemotherapy regimen (%) | — | 0 | — | 54 | — | 0 |
| Received intended CAR T cell (%) | 94 | — | 96 | — | 97.8 | — |
| Median time to CAR T-cell infusion in days, (interquartile range* or range†) | 29 (27-34)* | — | 52 (31-135)† | — | NR | — |
| Received intended ASCT (%) | — | 36 | — | 32.5 | — | 45.6 |
| Crossover pn protocol (%) | — | — | — | 51 | — | 51 |
| Received cellular therapy off protocol (%) | — | 56 | — | — | — | |
| Follow up, median in months | 24.9 | 10 | 6.2 | |||
| ORR/CR rate (%) | 83/65 | 50/32 | 46/28 | 43 /28 | 86/66 | 48/39 |
| EFS, median in months | 8.3 | 2 | 3 | 3 | 10.1 | 2.3 |
| EFS, % (timepoint in months) | 41 (24 mo) | 16 (24 mo) | NR | NR | 63 (6 mo) | 33 (6 mo) |
| EFS HR (95% CI) | 0.4 (0.31-0.51) | 1.07 (0.82-1.4) | 0.35 (0.23-0.53) | |||
| PFS, median in months | 14.7 | 3.7 | NR | NR | 14.8 | 5.7 |
| PFS HR (95% CI) | 0.49 (0.37-0.65) | NR | 0.406 (0.21-0.66) | |||
| OS, median in months | NE | 25.7 | 16.9 | 15.3 | NE | 16.4 |
| OS HR (95% CI) | 0.708 (0.515-0.972)‡ | NR | 0.51 (0.26-1.004) | |||
NE, not evaluable; NR, not reported; SOC, standard of care.
Interquartile range of time from leukapheresis to infusion of axi-cel.
Range of time from leukapheresis to infusion of tisa-cel.
Updated data from reference 42.
The ZUMA-7 trial enrolled 359 patients with a median age of 59 years (maximum age, 81 years).1 Baseline characteristics were well balanced, with >70% of patients having primary refractory disease. As per protocol, no patients on the axi-cel arm received bridging chemotherapy, but 36% received corticosteroids to control disease-related symptoms. Ninety-four percent of patients received the intended CAR T-cell therapy, with median time from randomization to infusion of 29 days. In contrast, only 36% of patients in the standard of care arm underwent ASCT. The overall response rate (ORR) and complete response (CR) rate were significantly greater in the axi-cel arm (83% and 65%) in comparison with the standard of care arm (50% and 32%). With a median follow-up time of 24.9 months, the median EFS was 8.3 months in the axi-cel arm vs 2 months in the standard of care arm and was 41% vs 16% at 24 months. Although the trial did not allow crossover, 56% of patients on the standard of care arm received subsequent cellular therapy off protocol. Based on the hierarchical statistical plan, OS was not significantly improved at the time of interim analysis, although a trend in favor of axi-cel compared with standard of care was observed (hazard ration [HR], 0.708; 95% confidence interval [CI], 0.515-0.972; P = .0159; median not reached vs 25.7 months).42
The Belinda trial enrolled 322 patients with a median age of 59.5 and 58 years in the tisa-cel and standard of care arms, respectively, and a maximum age of 79 years.2 Baseline characteristics were well balanced, with >65% of patients having primary refractory disease. Bridging chemotherapy was given to 83% of patients on the tisa-cel arm, with 48% receiving >1 cycle and 12% receiving >1 regimen. At the 6-week assessment, 26% of patients had progressive disease in the tisa-cel arm (prior to infusion) compared with 14% in the standard of care arm. Nonetheless, 95.7% of patients received the intended CAR T-cell therapy, although median time from randomization to infusion of tisa-cel was quite prolonged at 52 days. Patients on the standard of care arm were required to undergo 2 salvage regimens before crossover to tisa-cel for inadequate response. As such, 54% of standard of care patients received 2 salvage regimens but only 32.5% received ASCT, of which 31% did so only after responding to the second salvage regimen. The ORR and CR rate were not different between the tisa-cel (46% and 28%) and standard of care arms (43% and 28%). With a median follow-up of 10 months, the median EFS of 3 months was similar in both arms (P = .61). In total, 51% of patients on the standard of care arm crossed over to receive tisa-cel on study. Because of the hierarchical statistical plan, OS was not formally tested but was numerically similar between the tisa-cel and standard of care arms (median, 16.9 vs 15.3 months).
The Transform trial enrolled 184 patients with a median age of 60 and 58 years in the liso-cel and standard of care arms, respectively, and a maximum age per protocol of 75 years.3 Baseline characteristics were well balanced, with >70% of patients having primary refractory disease. One cycle of bridging chemotherapy was given to 63% of patients on the liso-cel arm, and 97.8% received the intended CAR T-cell therapy (median infusion time not reported to date). In contrast, only 45.6% of patients on the standard of care arm received ASCT. The ORR and CR rate were significantly greater in the liso-cel arm (86% and 66%) in comparison with the standard of care arm (48% and 39%). With a short median follow-up time of 6.2 months at a prespecified interim analysis, the median EFS was 10.1 months in the liso-cel arm vs 2.3 months in the standard of care arm and was 63% vs 33% at 6 months. In total, 51% of standard of care patients crossed over to liso-cel on study. The interim analysis of OS did not demonstrate a statistical improvement to date; however, there was a trend to improvement in the liso-cel arm (HR, 0.509; 95% CI, 0.258-1.004; P = .0257; median not evaluable [NE] vs 16.4 months). At the time of this review, the Transform trial has only been presented in abstract form.
Grade ≥3 toxicities were seen in nearly 90% of patients on both arms of all 3 studies; however, key differences included CRS and neurologic toxicities in the CAR T-cell arms and more frequent cytopenias and infections in the ASCT arms1-3 (Table 4). Toxicities of interest in the CAR T-cell therapy arms were similar to rates reported within the respective pivotal trials.15-17 Rates of grade ≥3 CRS were relatively low in all trials (1%-6%). ICANS was more common in ZUMA-7, with grade ≥3 toxicity in 21% of patients compared with 2% in Belinda and 4% in Transform. Additional follow-up is needed for long-term toxicities; however, ZUMA-7 reported grade ≥3 cytopenias 30 days from definitive therapy in 29% of axi-cel–treated patients in contrast with 19% of patients who received ASCT. Toxicities observed to be more common in the standard of care arm than in the CAR T-cell therapy arm included febrile neutropenia, anemia, and nausea. Importantly, the fatal adverse event rate attributable to therapy was not different between the arms in any of the trials. Quality of life, measured with patient-reported outcomes, was improved in the CAR T-cell arms of ZUMA-7 and Transform.43,44
Adverse events of interest
| ZUMA-7 | Belinda | Transform | ||||
|---|---|---|---|---|---|---|
| Axi-cel | SOC | Tisa-cel | SOC | Liso-cel | SOC | |
| CRS, any grade (%) | 92 | — | 61 | — | 49 | — |
| CRS, grade ≥3 (%) | 6 | — | 5 | — | 1 | — |
| Neurologic toxicity, any grade (%) | 60 | — | 10 | — | 12 | — |
| Neurologic toxicity, grade ≥3 (%) | 21 | — | 2 | — | 4 | — |
| Tocilizumab use (%) | 65 | — | 32 | — | 24 | — |
| Corticosteroid usage for toxicity management (%) | 24 | — | 10 | — | 17 | — |
| Anemia, grade ≥3 (%) | 30 | 39 | 33 | 58 | 49 | 49 |
| Thrombocytopenia, grade ≥3 (%) | 15 | 57 | 32 | 48 | 49 | 64 |
| Neutropenia, grade ≥3 (%) | 69 | 41 | 40 | 39 | 80 | 51 |
| Febrile neutropenia, grade ≥3 (%) | 2 | 27 | 13 | 25 | 15 | 24 |
| Fatigue, any grade (%) | 42 | 52 | 24 | 31 | 39 | 38 |
| Nausea, any grade (%) | 41 | 69 | 41 | 49 | 53 | 57 |
| ZUMA-7 | Belinda | Transform | ||||
|---|---|---|---|---|---|---|
| Axi-cel | SOC | Tisa-cel | SOC | Liso-cel | SOC | |
| CRS, any grade (%) | 92 | — | 61 | — | 49 | — |
| CRS, grade ≥3 (%) | 6 | — | 5 | — | 1 | — |
| Neurologic toxicity, any grade (%) | 60 | — | 10 | — | 12 | — |
| Neurologic toxicity, grade ≥3 (%) | 21 | — | 2 | — | 4 | — |
| Tocilizumab use (%) | 65 | — | 32 | — | 24 | — |
| Corticosteroid usage for toxicity management (%) | 24 | — | 10 | — | 17 | — |
| Anemia, grade ≥3 (%) | 30 | 39 | 33 | 58 | 49 | 49 |
| Thrombocytopenia, grade ≥3 (%) | 15 | 57 | 32 | 48 | 49 | 64 |
| Neutropenia, grade ≥3 (%) | 69 | 41 | 40 | 39 | 80 | 51 |
| Febrile neutropenia, grade ≥3 (%) | 2 | 27 | 13 | 25 | 15 | 24 |
| Fatigue, any grade (%) | 42 | 52 | 24 | 31 | 39 | 38 |
| Nausea, any grade (%) | 41 | 69 | 41 | 49 | 53 | 57 |
SOC, standard of care.
Discussion
The ZUMA-7, Belinda, and Transform trials all had the same objective: to determine whether CAR T-cell therapy would improve outcomes in patients with high-risk R/R LBCL to supplant the longstanding standard of care of platinum-based salvage followed by ASCT. Results yielded discordant findings, with ZUMA-7 and Transform meeting their primary end point of improved EFS and Belinda demonstrating no difference in outcome between strategies. How can these disparate findings be resolved to guide treating clinicians? Although the 3 trials had numerous similarities, important differences in trial design, including the allowance of bridging therapy, the definition of EFS, and the potential for crossover, likely had major implications on overall results.
Because of trial differences, direct cross-trial comparisons are not feasible. However, it is important to consider whether comparable patients were enrolled and whether patients are representative of those seen in routine practice. Despite similar inclusion criteria, ZUMA-7 did not allow bridging chemotherapy, precluding participation of patients requiring immediate treatment. Real-world studies indicate approximately 50% of patients receive bridging therapy.29,30 This exclusion may have introduced a selection bias in favor of patients with less aggressive biology. Bridging therapy has been associated with inferior outcomes in patients treated with CAR T-cell therapy, as it is mainly used in patients with poorly controlled disease.29,30,37,38 Lymphoma tumor burden and aggressiveness as measured by elevation in lactate dehydrogenase, number of extranodal sites, and total metabolic tumor volume are associated with poorer outcomes.29,30,45 Comparison of clinical parameters between trials is difficult, as lactate dehydrogenase, extranodal sites, and disease bulk were not universally reported, and ZUMA-7 and Transform reported age-adjusted international prognostic index, whereas Belinda did not. Median age was similar across trials, despite the age restriction of 75 years within Transform. A slight female predominance within the CAR T-cell arms of Transform and ZUMA-7 occurred, which may be relevant as female patients have improved outcomes with immunotherapy46 and CAR T-cell therapy.29 All 3 trials had a similar proportion of patients with primary refractory disease and high-grade B-cell lymphoma. Cell-of-origin data have not been reported for the Transform study, but Belinda had a higher percentage of activated B-cell patients (32%) than ZUMA-7 (9%), although the prognostic value of cell-of-origin is uncertain in patients with R/R disease and was not associated with differences in EFS rates in the Belinda trial.47,48 It is reassuring that the positive findings observed in ZUMA-7 were consistent with those observed in the Transform trial, where bridging chemotherapy was used in 63% of liso-cel patients, representing a more real-world approach. However, it should be noted that the highest use of bridging chemotherapy (83%) was reported in Belinda, with 48% receiving more than 1 cycle, possibly indicating patients with more aggressive and/or chemotherapy-insensitive disease.
Another major difference between trials was the definition of EFS. All trials included disease progression and death as an event, as well as lack of a complete or partial response (stable disease) at a designated time point. This time point differed between trials, being longest in ZUMA-7 at 150 days, compared with only 9 weeks in Transform and 12 weeks in Belinda. Most impactful was the requirement of a second salvage regimen within the Belinda trial before crossover to tisa-cel, which was not considered an event, whereas such initiation of new antilymphoma therapy would have been considered an event in both ZUMA-7 and Transform. Thus, patients in the standard of care arm of Belinda were given a second chance to respond to salvage, which led to 54% of patients receiving 2 salvage regimens, representing 31% of patients proceeding to ASCT. Despite this allowance, the Belinda trial had the lowest percentage of patients proceeding to ASCT at 32.5%. Had only 1 salvage regimen been required, less than 25% of patients would have received ASCT in comparison with 36% on ZUMA-7 and 45.6% on Transform, possibly reflecting a higher-risk cohort within Belinda.
The ability to deliver the intended CAR T-cell therapy was similarly high in all trials, ranging from 94% to 97.8%. However, the time from randomization to CAR T-cell infusion, which was not reported in Transform, was markedly prolonged in Belinda (median, 52 days) compared with the ZUMA-7 trial (29 days). This difference, largely because of manufacturing considerations, may have negatively impacted CAR T-cell outcomes in Belinda. First, the infusion delay may have led to increased tumor burden, as evidenced by 26% of patients having progressive disease at the week 6 response assessment, limiting CAR T-cell utility. Ultimately, 83% of patients on the tisa-cel arm required bridging therapy, with 12% receiving 2 different bridging regimens, effectively receiving CAR T-cell therapy as third-line treatment. Finally, because stable disease at week 12 was considered an event, in light of the previously reported median response time of 2 months,16 the delayed infusion may not have permitted sufficient time for adequate response assessment. Based on trial differences, the comparative efficacy of CAR T-cell products cannot be directly inferred from these results. However, the possibility of lower efficacy of tisa-cel in the second-line setting cannot be excluded.
In addition to a marked improvement in EFS, both ZUMA-7 and Transform trials demonstrated a significant improvement in PFS. With a median follow-up of 24.9 months, ZUMA-7 provides the most mature follow-up and demonstrates a remarkable 2-year PFS of 46% within the axi-cel arm. The 2-year PFS of 27% in the standard of care arm highlights the limitation of relying on further cytotoxic therapy in this population of patients with inherent chemotherapy resistance. Although Transform was reported as a planned interim analysis with limited follow-up, more than two thirds of patients in the standard of care arm have already experienced an event indicating that the findings are unlikely to change. Assessment of OS benefit at the interim analyses remains immature; however, there is a trend toward improved OS within ZUMA-7 and Transform. This suggests that third-line CAR T-cell therapy following the failure of salvage therapy may not fully compensate for the benefit achieved with CAR T-cell therapy in the second-line setting. The OS on the standard of care arms was highest in ZUMA-7 with a median of 25.7 months,42 compared with 16.4 months in Transform and 15.3 months in Belinda, and with the caveat of differences in follow-up, could suggest a baseline difference in the populations enrolled.
One consistent finding within all 3 trials is the dismal outcome observed with planned salvage chemotherapy and ASCT in this poor-risk population. The similar low rates of transplantation ranging from 32.5% to 45.6% are in keeping with previous expectations, and few durable responses were seen.12,13,49 It is noteworthy that in all 3 trials, a higher percentage of patients (51%-56%) within the standard of care arms received CAR T-cell therapy than ASCT, a number that may increase with longer follow-up as additional patients relapse and will undoubtedly confound assessment of OS. It has been questioned whether a more appropriate design of these trials would have been to treat all patients with salvage chemotherapy and only randomize patients with a response, given the resource and cost implications of CAR T-cell therapy. In view of the lack of predictive markers for chemo-sensitivity, the low expectation of response to salvage therapy, and poor anticipated outcomes in this high-risk population, the goal of these trials was to investigate the alternative option of immediate CAR T-cell therapy. Exposing all patients to salvage therapy first would incur needless toxicity in most patients and inevitably delay the administration of CAR T-cell therapy in all patients, which may undermine its value. Available data suggest that patients with greater exposure to prior therapy may have less potent CAR T-cell products.50 The ZUMA-7 and Transform trials demonstrate the superiority of second-line CAR T-cell therapy in this intention-to-treat high-risk population.
The results from the ZUMA-7 and Transform studies justify a paradigm shift to second-line CAR T-cell therapy (axi-cel or liso-cel) in suitable patients with LBCL who are refractory to or relapse within 1 year of completion of first-line therapy. The negative results observed in the Belinda trial likely reflect the limitation of CAR T-cell therapy when administered in a delayed fashion or in the setting of poorly controlled disease. These results should not be extrapolated beyond the study population as patients with later relapse can have reasonable outcomes following ASCT, and it is likely the mechanisms of resistance may differ according to timing of relapse.8,14 A recent population-based review of outcomes in transplant-eligible patients according to timing of relapse, demonstrated a 5-year PFS of 12% with platinum-based salvage followed by ASCT in patients who were primary refractory or relapsed within 12 months of initial diagnosis compared with a 5-year PFS of 49% in patients who relapsed beyond 2 years.51 Whether patients with refractory or early relapsing LBCL who derive benefit from bridging chemotherapy started urgently before leukapheresis should be reconsidered for ASCT cannot be determined based on trial design. Importantly, patients with extensive poorly controlled disease prior to CAR T-cell infusion may benefit less from this approach, and proceeding with CAR T-cell treatment should be weighed against other available options.
The current trials were restricted to a transplant-eligible population, although 2 trials had no upper age limit. The criteria for transplant eligibility vary between institutions and are often based on performance status, physiologic age, organ function, and tolerance of first-line therapy. In view of the favorable efficacy and toxicity profile, it could be argued that second-line CAR T-cell therapy should be offered to some patients previously considered transplant ineligible. The population of CAR T-cell–eligible patients is somewhat larger than the transplant-eligible population, with feasibility demonstrated in older patients and those with modest comorbidities, albeit with somewhat higher toxicity.29-33 The ongoing PILOT study is evaluating the safety and efficacy of liso-cel as second-line therapy for transplant-ineligible patients with aggressive B-cell lymphoma, with preliminary results demonstrating similar safety and efficacy to the phase 2 pivotal trial.52 However, use of CAR T-cell therapy in high-risk transplant-ineligible patients should incorporate a careful consideration of the risk of toxicity and other treatment options.
A proposed management algorithm for second-line therapy of R/R LBCL is shown in Figure 2, with projected outcomes based on historic expectations and data from phase 3 CAR T-cell therapy trials. Although longer-term results and safety information will be required to fully assess the merit of second-line axi-cel or liso-cel therapy, the 2-year data from ZUMA-7 are reassuring as few patients achieving CR within the pivotal trials relapsed after 1 year.25 In the absence of a definitive OS benefit (which may emerge over time), cost-effectiveness analyses will be helpful to support second-line CAR T-cell therapy but should take into account the downstream treatment course of ASCT, including crossover to CAR T-cell therapy and use of costly novel agents. It remains unknown whether patients who progress or relapse despite receiving CAR T-cell therapy in the second-line setting would benefit from ASCT, the potential cost of which should also be considered. Further insight regarding outcomes within histologic, biological, and clinical subgroups and mechanisms of resistance are also required to ensure patient selection is optimized. Identification of better bridging strategies with novel agents will be important to broaden eligibility and enhance outcomes. Future advances, including improvements in CAR T-cell manufacturing, novel CAR T-cell constructs, and off-the-shelf or allogeneic cell therapies are under investigation and may further improve outcomes.
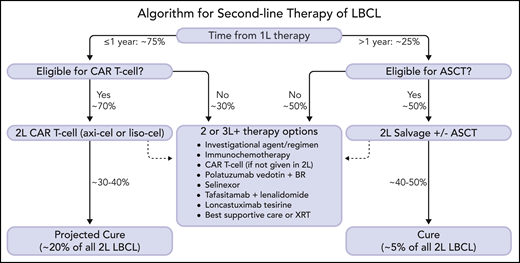
A new treatment algorithm for patients with R/R LBCL after first-line therapy is proposed. Patients with refractory or relapsed disease within 1 year of completion of initial therapy should be considered for second-line CAR T-cell therapy (axi-cel or liso-cel) if eligible. Patients who relapse >1 year from initial therapy should be considered for ASCT if transplant-eligible. Patients who are not eligible for second-line cellular therapy have numerous therapeutic alternatives, including investigational agents/clinical trials. Selection of therapy should be individualized based upon disease and patient characteristics, treatment goals, patient preference, and logistical factors (some agents may only be indicated for third-line therapy and beyond). At the time of publication, no CAR T-cell therapy has yet been approved by the regulatory agencies for second-line therapy, but regulatory review is ongoing. Percentages are estimated and projected based upon data from clinical trials and historical outcomes. The dashed arrows indicate treatment path for patients who are not cured by cellular therapy. 1L, first-line; 2L, second-line; 3L+, third-line and beyond; ASCT, autologous stem cell transplantation; axi-cel, axicabtagene ciloleucel; BR, bendamustine and rituximab; CAR T-cell, chimeric antigen receptor T-cell therapy; LBCL, large B-cell lymphoma; liso-cel, lisocabtagene maraleucel; XRT, radiation therapy. Professional illustration by Somersault18:24.
A new treatment algorithm for patients with R/R LBCL after first-line therapy is proposed. Patients with refractory or relapsed disease within 1 year of completion of initial therapy should be considered for second-line CAR T-cell therapy (axi-cel or liso-cel) if eligible. Patients who relapse >1 year from initial therapy should be considered for ASCT if transplant-eligible. Patients who are not eligible for second-line cellular therapy have numerous therapeutic alternatives, including investigational agents/clinical trials. Selection of therapy should be individualized based upon disease and patient characteristics, treatment goals, patient preference, and logistical factors (some agents may only be indicated for third-line therapy and beyond). At the time of publication, no CAR T-cell therapy has yet been approved by the regulatory agencies for second-line therapy, but regulatory review is ongoing. Percentages are estimated and projected based upon data from clinical trials and historical outcomes. The dashed arrows indicate treatment path for patients who are not cured by cellular therapy. 1L, first-line; 2L, second-line; 3L+, third-line and beyond; ASCT, autologous stem cell transplantation; axi-cel, axicabtagene ciloleucel; BR, bendamustine and rituximab; CAR T-cell, chimeric antigen receptor T-cell therapy; LBCL, large B-cell lymphoma; liso-cel, lisocabtagene maraleucel; XRT, radiation therapy. Professional illustration by Somersault18:24.
Authorship
Contribution: J.W. and L.H.S. analyzed the data and wrote the paper.
Conflict-of-interest disclosure: J.W. has received research funding from Kite/Gilead, Novartis, BMS, Genentech, AstraZeneca, Janssen, Morphosys/Incyte, Precision Biosciences, and ADC Therapeutics and has received consulting funding from Kite/Gilead, Novartis, BMS, Janssen, AstraZeneca, Genentech, Morphosys/Incyte, ADC Therapeutics, Iksuda, Umoja, MonteRosa, and Merck. L.H.S. has received advisory board fees from AbbVie, AstraZeneca, Gilead, Genentech, Janssen, Merck, Takeda, Apobiologix, Acerta, Celgene, Kite, Karyopharm, Morphosys, Lundbeck, TG Therapeutics, Verastem, Sandoz, Incyte, Novartis, Genmab, and Debiopharm; grant support and advisory board fees from Teva; advisory board fees and lecture fees from Roche and Seattle Genetics; research funding from Teva and Roche/Genentech; and consulting funding from Abbvie, Acerta, Amgen, Apobiologix, Astra Zeneca, Celgene, Chugai, Gilead, Incyte, Janssen, Kite, Karyopharm, Lundbeck, Merck, Morphosys, Roche/Genentech, Sandoz, Seattle Genetics, Servier, Teva, Takeda, TG Therapeutics, and Verastem.
Correspondence: Jason Westin, 1515 Holcombe Blvd, Unit 0429, Houston, TX 77030; e-mail: jwestin@mdanderson.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal