

In this issue of Blood, Antoszewski et al report that the transcription factor T-cell factor 1 (Tcf1) remodels chromatin in Notch-activated early hematopoietic progenitor cells to promote transformation through oncogenes like MYC.1
In 1957, Francis Crick put forth the central dogma of information flowing from DNA to RNA. Since then, we have learned that this process, called transcription, is tightly regulated in highly complicated structures. In the current model of transcription, sequence-specific transcription factors bind DNA, forming macromolecular complexes that displace nucleosomes at promoter and enhancer elements. Next, chromosomal looping brings enhancers into proximity of the transcriptional start site. These enhancer-promoter interactions are restricted within giant folds of chromatin called topologically associating domains (TADs). Finally, RNA polymerase binds and initiates transcription. Since these transcriptional structures are so intricate, developing cancer cells often find it easier to transform from normal cells by putting these structures into overdrive rather than creating new ones from scratch. A classic example are the structures created by supraphysiological Notch signaling in T-cell acute lymphoblastic leukemia (T-ALL). During normal T-cell development, ligands on stromal cells activate Notch1 receptors on the surface of hematopoietic precursors to promote T-cell commitment and robust expansion (see figure).2 But unfortunate events like chromosomal translocations or mutations can raise Notch signaling to hyperactive levels, inducing overexpression of oncogenic target genes like MYC through native promoter-enhancer structures.3,4 Normal T-cell precursors are transformed into malignant T-ALL, a devastating outcome.
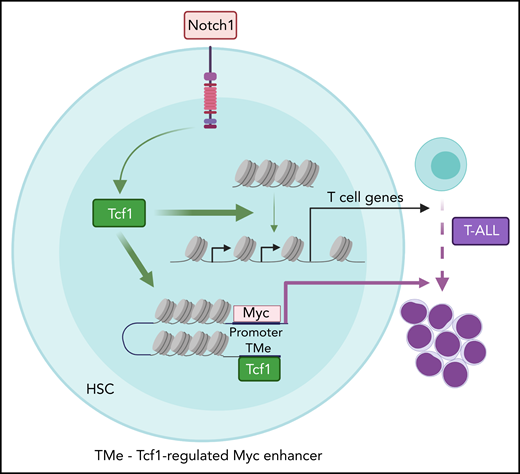
Model depicting the genome-scale actions of T-cell factor 1 (Tcf1) in promoting chromatin accessibility for T-cell development and leukemogenesis. Physiological Notch signaling directs Tcf1 to displace nucleosomes, paving the way for additional transcription factors to bind and induce T-cell differentiation genes. In contrast, supraphysiological Notch signaling directs Tcf1 to open a newly appreciated myelocytomatosis protooncogene (Myc) enhancer (TMe) that transforms Notch actions from physiological to oncogenic. HSC, hematopoietic stem cell. See the visual abstract in the article by Antoszewski et al that begins on page 2483.
Model depicting the genome-scale actions of T-cell factor 1 (Tcf1) in promoting chromatin accessibility for T-cell development and leukemogenesis. Physiological Notch signaling directs Tcf1 to displace nucleosomes, paving the way for additional transcription factors to bind and induce T-cell differentiation genes. In contrast, supraphysiological Notch signaling directs Tcf1 to open a newly appreciated myelocytomatosis protooncogene (Myc) enhancer (TMe) that transforms Notch actions from physiological to oncogenic. HSC, hematopoietic stem cell. See the visual abstract in the article by Antoszewski et al that begins on page 2483.
T-ALL is an aggressive hematological malignancy, accounting for ∼15% of pediatric ALL and ∼25% of adult T-ALL cases. Adult T-ALL show only ∼50% long-term survival, whereas children fare better but are at high risk for long-term toxicities. To improve outcomes, investigators initially sought to inhibit Notch signaling with drugs. However, early clinical trials showed that pan-Notch inhibitors were too toxic. Because of this, investigators turned to identifying target genes of Notch in hopes of finding more attractive therapeutic targets. Although these studies revealed T-ALL drivers important for diverse cancerous functions, it remained unclear which target genes of Notch can remodel chromatin to set the stage for T-ALL transformation. Notch itself does not have this function. With the emergence of protein-protein interaction inhibitors, protein degraders, and covalent inhibitors in the modern era, transcriptional regulators are no longer thought of as “undruggable.” In this context, Antoszewski et al reveal the key role played by the transcription factor Tcf1, which is encoded by the Notch target gene Tcf7, in shaping chromatin architecture to facilitate initiation of Notch-induced T-ALL.
Antoszewski et al zeroed in on Tcf1 because it has “pioneer” activity, which is the ability to open repressive chromatin to allow other transcription factors to bind.5 During early T-cell development, Notch signaling directly induces Tcf1 expression, which establishes an epigenetic landscape that promotes expression of genes important for T-cell fate.5-7 To test whether Tcf1 is also important for T-ALL initiation, Antoszewski et al conditionally deleted Tcf7 in a genetically engineered mouse model of Notch-induced T-ALL. T-ALL initiation was abrogated. In contrast, deletion of the gene encoding β-catenin, a well-known Tcf1 cofactor in the Wnt pathway, had less impressive effects. Thus, Tcf1 primarily functions in a β-catenin/Wnt-independent manner. To determine the mechanism, Antoszewski et al applied a comprehensive suite of genomic tools to analyze chromatin 3D architecture, chromatin accessibility, enhancer activity, and gene expression in Notch-activated mouse bone marrow hematopoietic progenitor cells. Tcf7 loss in these rare cells reversed many Notch-induced changes in chromatin accessibility and T-cell specific gene expression. Tcf7 loss also reversed some effects of Notch on TAD boundaries and enhancer-promoter interactions, which often correlated with effects on gene expression and enhancer activity. Next, Antoszewski et al focused on a Notch-dependent distal Myc enhancer region that is known to drive T-ALL transformation and maintenance. Tcf7 loss not only rendered this region inaccessible in Notch-activated cells but also another enhancer ∼14 kb more distal, which they nicked named “TMe” for Tcf1-regulated Myc enhancer. To establish the importance of this enhancer, they deleted the TMe in their mouse models. Strikingly, TMe deletion disabled Notch-induced T-cell leukemogenesis while preserving Notch-induced T-cell development.
The Antoszewski et al study merges high quality and elegant mouse modeling with comprehensive and technically challenging differential analysis of chromatin structure in rare hematopoietic progenitor cells. These data combined with previous literature strongly implicate Tcf7 as an important Notch target gene that shapes the chromatin environment for other transcription factors to bind and intensify oncogene expression. Unfortunately, the lack of a high-quality Tcf1 antibody for chromatin immunoprecipitation sequencing makes it difficult to separate direct vs indirect effects as downstream targets of Tcf1, like Gata3, have chromatin remodeling activity.8 It also remains unclear whether transforming cells stimulated by weaker Notch signals, such as through Notch ligand-receptor interactions, access oncogenic regulatory regions through similar mechanisms. Given its importance as a primary regulator of chromatin accessibility in early T cells,5 Tcf1 likely plays a role even in such cases. It is also unclear whether Tcf1 maintains chromatin architecture in established T-ALL or becomes superseded by other factors like Gata3.8 Other groups showed that Notch signaling itself has limited activity in this regard in cancer.9,10 Finally, it is unclear how Notch1/Tcf1 mechanisms at native cis-elements interact with a dysregulated sea of aberrancies in transcriptional regulators, DNA methylation, and mutations in promoters, enhancers, and insulators during human T-ALL initiation and maintenance. Future studies will be needed.
In an impressively ambitious and resource-heavy undertaking, Antoszewski et al rewards us with a genome-scale understanding of how Notch and Tcf1 transform chromatin into a nurturing environment for the development of T-cell leukemia. We have learned that Notch signaling directs Tcf1 to evict nucleosomes from repressed chromatin to recruit oncogenic transcription factors that fire up enhancers important for T-cell commitment and expansion (see figure). We have even seen that a previously unrecognized MYC enhancer (TMe) can separate oncogenic from physiological Notch functions, which might be key to targeting Notch without intolerable side effects. By dissecting the interactions at these oncogenic enhancers, we might find safe ways to quench their activities and reprogram malignant T cells back to their normal selves.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal