

In this issue of Blood, Kermasson et al report the discovery of biallelic Apollo/DCLRE1B mutations in 3 unrelated children who presented with hematologic, immunologic, and mucocutaneous features of dyskeratosis congenita, albeit with normal-length telomeres.1 Using a series of biochemical and genetic experiments, they showed that mutations affect both the DNA repair and telomere biology function of the Apollo gene.
Apollo, the son of Zeus and Leto, and twin brother of Artemis, the most admired of Greek gods, was the patron of the music, dance, poetry, archery, light, and healing and the protector of the youth. It comes therefore as no surprise that the DCLRE1B (SNM1B) gene was named Apollo, given its multifaceted role as an exonuclease in DNA repair and a protector of telomeres during the S phase. The actual etymologic origin comes from the initial observation that the Apollo/DCLRE1B gene shares high sequence similarity with Artemis/DCLRE1C, a paralog protein involved in DNA repair with a key role in B- and T-cell development.2 Both genes belong to a superfamily of nucleases containing metallo-β-lactamase and β-CASP domains, which together exert nuclease activity. Apollo is necessary for the normal cellular response to DNA interstrand crosslinks (ICLs); is involved in both ATM- and ATR-mediated DNA damage signaling and the repair of stalled replication forks; is linked to the FA pathway (physically interacting with FANCD2 and FANCP); and finally, is the protector of telomeres, acting as a shelterin accessory protein and vitally contributing to the generation and maintenance of telomeric overhangs.3
How does Apollo fit into the family of genes associated with telomeropathies (see figure)? In trying to explain this, one must first embark on a semantic journey into the past. The entity dyskeratosis congenita, the name first coined by dermatologists over a century ago and later adopted by pediatricians, is an inherited bone marrow failure (BMF) syndrome characterized by the triad of oral leukoplakia, nail dystrophy, and reticular skin pigmentation, often with other premature aging phenotypes related to the underlying telomere shortening.4 However, not all patients present with the typical triad, and the only manifestation may be BMF, pulmonary fibrosis, or cancer, particularly in adults.5 To address the nomenclature issue, many investigators therefore prefer the expression short telomere syndrome. However, some patients have only a mild (∼1st to 10th percentile) reduction in telomere length. Moreover, mutations in the telomere biology gene POT1 are associated with increased telomere length and CTC1 with ambiguous telomere length. Here, Apollo provides another puzzle piece, representing a prototypic telomere biology disorder (TBD) that manifests with classic dyskeratosis congenita features, albeit normal blood telomere length. TBD is now increasingly used as an umbrella term for all syndromes with abnormal telomere biology and allows investigators to think outside the box when addressing such contradictions. The normal telomere length in Apollo deficiency is an important take-home message.
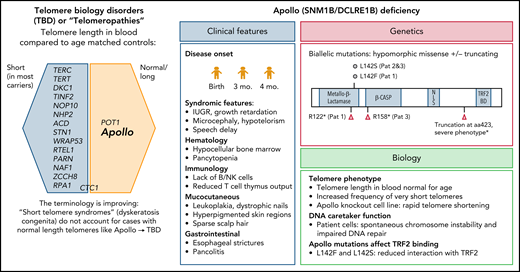
Genetic spectrum of telomeropathies and characteristics of Apollo deficiency. The genes identified thus far as being associated with human telomeropathies (left). Clinical, genetic, and biological features of Apollo deficiency (right). IUGR, intrauterine growth restriction; mo, months; Pat, patient. *Touzot et al.6
Genetic spectrum of telomeropathies and characteristics of Apollo deficiency. The genes identified thus far as being associated with human telomeropathies (left). Clinical, genetic, and biological features of Apollo deficiency (right). IUGR, intrauterine growth restriction; mo, months; Pat, patient. *Touzot et al.6
Kermasson et al identified 3 unrelated patients who shared the unique phenotype of early-onset hypocellular BMF with B- and NK-cell immunodeficiency. This report is a long-awaited addition to the first and only case of an Apollo splice site alteration and severe dyskeratosis congenita phenotype (with similar clinical features and also normal telomere length), previously reported by Touzot et al.6 In the current cohort of Kermasson et al, the hematologic and immunologic problems manifested at a median age of 3 months (range, 0-4), with mucocutaneous and gastrointestinal abnormalities emerging later, at a median age of 5 years (range, 0.8-8). The patients carried either hypomorphic missense mutations affecting highly conserved residue L142 or truncating loss-of-function mutations (most likely resulting in truncated and unstable protein).
It is interesting that the patients’ parents, who were heterozygous mutation carriers were clinically healthy. This is similar to other DNA repair disorders like Fanconi anemia, where heterozygous mutation carriers generally have no increased disease risk and suggests sufficient compensatory capacity of the remaining wild type allele.
It is well known that Apollo interacts with the TRF2 component of the shelterin complex and protects telomeres during or after replication.7 The missense mutations expressed in a cell line resulted in a decrease in binding to TRF2. Although patient blood and fibroblast cells had no significant decrease in global telomere length compared with controls, using a sensitive technique called a telomere shortest length assay applied to blood cells of 1 patient, Kermasson et al found that the frequency of very short telomeres was increased. This effect may be explained by reduced availability of a telomeric G-overhang, and in fact they found a 30% to 50% reduction in the G-overhang length, supporting that patients’ mutations affect the nuclease activity of Apollo. Of note, when Apollo was knocked out in an HT1080 telomerase–positive cell line, Kermasson et al observed rapid reduction of global telomere length that contrasted with normal telomere length in patient cells. One could hypothesize that patients’ mutations are hypomorphic and retain some activity of Apollo, thus not translating to a global loss of telomere length, or that the telomere loss may be sudden and context or tissue dependent. This certainly leaves room for exciting future discoveries on the general role of Apollo in telomere biology.
Given the role of Apollo as DNA caretaker protein, it would be important to assess how these mutations affect the DNA repair function of Apollo. Using patient cells and a gene complementation system, Kermasson et al showed an impaired ICL repair, altered replicative stress response, and DNA double-strand–break repair. Further work is needed to pinpoint the exact mechanisms behind these phenotypes and the potential long-term clinical consequences.
In summary, this work establishes Apollo deficiency as a new BMF telomeropathy syndrome with textbook clinical features of severe dyskeratosis congenita, despite normal global telomere length.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal