


Abstract
Hairy cell leukemia (HCL) responds very well to frontline chemotherapy with purine analogs (cladribine and pentostatine). However, approximately half of patients experience 1 or more relapses, which become progressively resistant to these myelotoxic and immunosuppressive agents. At progression, standard therapeutic options include a second course of purine analogs alone or in combination with rituximab and, upon second relapse, therapy with the anti-CD22 immunotoxin moxetumomab pasudotox. Furthermore, blockade of the mutant BRAF-V600E kinase (the pathogenetic hallmark of HCL) through orally available specific inhibitors (vemurafenib or dabrafenib) effaces the peculiar morphologic, phenotypic, and molecular identity of this disease and its typical antiapoptotic behavior and is emerging as an attractive chemotherapy-free strategy in various clinical scenarios. These include patients with, or at risk of, severe infections and, in a highly effective combination with rituximab, patients with relapsed or refractory HCL. Other treatments explored in clinical trials are BTK inhibition with ibrutinib and co-inhibition of BRAF (through dabrafenib or vemurafenib) and its downstream target MEK (through trametinib or cobimetinib). Here, we focus on our experience with BRAF inhibitors in clinical trials and as off-label use in routine practice by presenting 3 challenging clinical cases to illustrate their management in the context of all available treatment options.
Introduction
Hairy cell leukemia (HCL) is a distinct indolent mature B-cell neoplasm,1 predominantly affecting males (male:female ratio, 4-5:1) with a median age of 55 to 60 years and usually presenting with cytopenias, few circulating leukemic cells, monocytopenia, and splenomegaly with no or little lymphadenopathy.1,2 HCL cells characteristically express B-cell antigens in addition to CD103, CD25, CD11c, cyclin D1, and annexin A1 (ANXA1).3 Moreover, HCL shows a distinct gene messenger RNA and microRNA expression profile4-6 and carries the BRAF-V600E mutation.7
HCL responds well to purine analogs (PAs) (cladribine, pentostatin) that induce durable complete remissions (CRs) in 85% to 90% of cases.2,8 In addition to the minority of poorly responding patients, in up to 58% of cases achieving CR the disease relapses within 5 years after initial treatment.9 Multiple relapses usually become progressively less sensitive to PAs10 that over time can cause cumulative myelotoxicity and profound immune suppression. PAs are indeed contraindicated in case of active infection, as well as moderate to severe hepatic and/or renal impairment. The most recent international guidelines suggest, at first relapse, retreatment with PAs if the previous remission lasted ≥5 years, with the addition of rituximab in case of a shorter first response.8 Nonmyelotoxic agents suitable for refractory/relapsed HCL are rituximab and interferon-α, but they usually produce few CRs.11,12 Moreover, interferon-α production has been even discontinued, although its pegylated formulation is available off-label.
During the past decade, much progress has been made in the treatment of refractory/relapsed HCL using moxetumomab pasudotox (Moxe)11,13,14 or BRAF inhibitors.15-17 Moxe is a recombinant immunotoxin containing an antibody fragment (Fv) directed against the B-cell molecule CD22 conjugated to a truncated Pseudomonas exotoxin A.13,18 In 77 patients with HCL (median of 3 previous therapies), Moxe resulted in 77% overall responses, with 43% CR and 35% MRD negativity rates19 (Table 1). At a median follow-up of 24.6 months, ∼45% of patients with HCL were in hematologic remission (HR; ie, resolution of cytopenias), and among patients achieving CR, the durability rate of HR was 83%.14 Approximately 10% of patients discontinued the drug due to reversible toxin-induced hemolytic uremic syndrome and capillary leak syndrome.20 In September 2018, Moxe was approved by the US Food and Drug administration for refractory/relapsed HCL after ≥2 prior therapies (of which ≥1 purine analog), with a boxed warning for hemolytic uremic syndrome and capillary leak syndrome. Unfortunately, after being approved also by the European Medicine Agency in February 2021, Moxe has been withdrawn from the European market by the pharmaceutical company and is not currently available outside the United States.
Efficacy of select standard and investigational treatments assessed in relapsed/refractory HCL
| HCL pts. | Response rate* | Cytopenia†-free survival rate at median follow-up | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study type | Drugs | Median prior therapies | Pts. refractory to a purine analog | OR | CR | MRD− | Time to neutrophils >1500/mm3 (median) | Time to platelets >10 0000/mm3 (median) | ||
| Case series10 | Purine analog + rituximab | 26 | 3 | 0% | 96% | 88% | Not reported | Not reported | Not reported | 87% at 78 mos |
| Phase-3 trial14,19 | Moxetumomab pasudotox | 77 | 3 | 49% | 77% | 43% | 35% | ≤4 wks | ≤4 wks | ∼45% at 24.6 mos |
| Phase-2 trials (n = 2)33 | Vemurafenib‡ | 28 | 3 | 21% | 89% | 32% | 0% | 4 wks | 2 wks | 23% at 23 mos |
| 26 | 3 | 4% | 92% | 38% | 0% | ≤4 wks | ≤4 wks | 68% at 12 mos | ||
| Case series (n = 2)31,32 | Low-dose vemurafenib§ | 21 | 3 | Not reported | 100% | 40% | Not reported | 7 wks | 4 wks | ≤50% at 17 mos |
| 27‖ | 3 | Not reported | 100% | 33% | Not reported | Not reported | Not reported | Not reported | ||
| Phase-2 trial35 | Vemurafenib¶ + rituximab | 30 | 3 | 37% | 87% | 87% | 60% | 4 wks | 2 wks | 78% at 37 mos |
| Phase-2 trial74 | Ibrutinib# | 28 | 4 | Not reported | 58% | 23% | ≤11% | 8 wks** | 20 wks | 43% at 42 mos |
| Phase-2 trial88 | Dabrafenib + trametinib†† | 43 | 3 | Not reported | 78% | 49% | 15% | Not reported | Not reported | 78% at 18 mos |
| HCL pts. | Response rate* | Cytopenia†-free survival rate at median follow-up | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study type | Drugs | Median prior therapies | Pts. refractory to a purine analog | OR | CR | MRD− | Time to neutrophils >1500/mm3 (median) | Time to platelets >10 0000/mm3 (median) | ||
| Case series10 | Purine analog + rituximab | 26 | 3 | 0% | 96% | 88% | Not reported | Not reported | Not reported | 87% at 78 mos |
| Phase-3 trial14,19 | Moxetumomab pasudotox | 77 | 3 | 49% | 77% | 43% | 35% | ≤4 wks | ≤4 wks | ∼45% at 24.6 mos |
| Phase-2 trials (n = 2)33 | Vemurafenib‡ | 28 | 3 | 21% | 89% | 32% | 0% | 4 wks | 2 wks | 23% at 23 mos |
| 26 | 3 | 4% | 92% | 38% | 0% | ≤4 wks | ≤4 wks | 68% at 12 mos | ||
| Case series (n = 2)31,32 | Low-dose vemurafenib§ | 21 | 3 | Not reported | 100% | 40% | Not reported | 7 wks | 4 wks | ≤50% at 17 mos |
| 27‖ | 3 | Not reported | 100% | 33% | Not reported | Not reported | Not reported | Not reported | ||
| Phase-2 trial35 | Vemurafenib¶ + rituximab | 30 | 3 | 37% | 87% | 87% | 60% | 4 wks | 2 wks | 78% at 37 mos |
| Phase-2 trial74 | Ibrutinib# | 28 | 4 | Not reported | 58% | 23% | ≤11% | 8 wks** | 20 wks | 43% at 42 mos |
| Phase-2 trial88 | Dabrafenib + trametinib†† | 43 | 3 | Not reported | 78% | 49% | 15% | Not reported | Not reported | 78% at 18 mos |
OR, overall response; CR, complete response; MRD-neg., negativity for minimal/measurable residual disease.
Among all patients starting study treatment.
Neutrophils <1500/mm3, platelets <10 0000/mm3, or hemoglobin <11 g/dL.
Nine hundred and sixty milligrams twice daily for a median of 16 and 18 weeks in the 2 trials (range 8-20 and 12-24, respectively).
For a median of 3 and 3.8 months in the 2 series (range, 1.9-8.9 and 1.7-19.9 months, respectively), largely at a dose of 240 mg twice daily or 480 mg twice daily.
Three out of 27 patients received dabrafenib plus or minus trametinib (not vemurafenib); 10/27 patients were included also among the 21 patients of the first series and were updated in the second series.
Nine hundred and sixty milligrams twice daily for a total of 8 weeks.
Four hundred and twenty milligrams or 840 mg continuously for an indefinite duration (median not reported; median follow-up, 3.5 years).
Refers to the lower threshold of 1000/mm3 (time to neutrophils >1500/mm3 was not reported).
One hundred and fifty milligrams twice daily (dabrafenib) plus 2 mg die (trametinib) continuously for an indefinite duration (median of 17 months).
The BRAF-V600E kinase mutation was discovered as the fundamental genetic event in HCL.7 It leads to constitutive activation of the MAPK pathway21 (Figure 1), and its pharmacologic blockade with specific BRAF inhibitors spoils leukemic cells of their unique molecular (Figure 1) and morphologic identity (trimming of hairy projections) before triggering their apoptosis22 (Figure 2). This breakthrough opened the way to a novel targeted therapy of this disease. Efficacy of BRAF inhibitors in refractory/relapsed HCL patients was demonstrated in anecdotical cases23-30 and in case series,31,32 mostly using low vemurafenib doses (240-480 mg twice daily), as well as in clinical trials33-35 using vemurafenib or dabrafenib at the standard dose (vemurafenib 960 mg twice daily and dabrafenib 150 mg twice daily, respectively) approved in BRAF-mutated metastatic melanoma.36-38 In particular, 2 phase-2 trials (in Italy and the USA) demonstrated a remarkable activity of vemurafenib, with 91% overall response rate and 35% CRs by intention-to-treat in 54 patients with refractory/relapsed HCL33 (Table 1). However, relapses were frequent, with a median relapse-free survival (RFS) from the end of treatment of 19 and 6 months after CR and partial remission (PR), respectively, in the Italian trial. Side effects, all reversible, were mostly of grade 1 to 2 and included skin manifestations (photosensitivity, rash, alopecia, palmar/plantar hyperkeratosis, warts), arthralgias or arthritis (usually responsive to low-dose steroids), pyrexia, and elevated serum bilirubin or lipase levels.

Constitutive activation of the RAS-RAF-MEK-ERK pathway by the BRAF-V600E mutation in HCL. (A) The RAS-RAF-MEK-ERK signaling pathway is physiologically triggered when a ligand binds to its surface receptor tyrosine kinase (RTK). This event, in turn, activates RAS and then BRAF (and CRAF, not shown). BRAF-CRAF heterodimers phosphorylate MEK (MEK1 and MEK2), which in turn phosphorylate ERK (ERK1 and ERK2). The BRAF-V600E mutation renders BRAF constitutively active independently from upstream signals and from heterodimerization with CRAF, thereby deregulating signaling activity. Active ERK enters the nucleus, where it triggers a transcriptional response (for example, through AP1 transcription factors). Such transcriptional response favors cell survival, proliferation, growth, and motility (as well as neoplastic transformation when deregulated by the BRAF-V600E mutation), and comprises genes that are generally induced by the RAF-MEK-ERK signaling pathway as well as genes induced by this cascade in the specific cellular context of HCL, including some of its diagnostic makers and the whole expression signature specific of this leukemia in comparison with normal mature B cells and other peripheral B-cell neoplasms.22 (B) HCL cells (brown) express the BRAF-V600E mutant protein (Ventana immunostaining with VE1 monoclonal antibody; immunoperoxidase and hematoxylin counterstain; ×400). (C) HCL cells express phospho-ERK (red) in the cytoplasm and the nucleus (anti–alkaline phosphatase immunostaining and hematoxylin counterstain; ×400).
Constitutive activation of the RAS-RAF-MEK-ERK pathway by the BRAF-V600E mutation in HCL. (A) The RAS-RAF-MEK-ERK signaling pathway is physiologically triggered when a ligand binds to its surface receptor tyrosine kinase (RTK). This event, in turn, activates RAS and then BRAF (and CRAF, not shown). BRAF-CRAF heterodimers phosphorylate MEK (MEK1 and MEK2), which in turn phosphorylate ERK (ERK1 and ERK2). The BRAF-V600E mutation renders BRAF constitutively active independently from upstream signals and from heterodimerization with CRAF, thereby deregulating signaling activity. Active ERK enters the nucleus, where it triggers a transcriptional response (for example, through AP1 transcription factors). Such transcriptional response favors cell survival, proliferation, growth, and motility (as well as neoplastic transformation when deregulated by the BRAF-V600E mutation), and comprises genes that are generally induced by the RAF-MEK-ERK signaling pathway as well as genes induced by this cascade in the specific cellular context of HCL, including some of its diagnostic makers and the whole expression signature specific of this leukemia in comparison with normal mature B cells and other peripheral B-cell neoplasms.22 (B) HCL cells (brown) express the BRAF-V600E mutant protein (Ventana immunostaining with VE1 monoclonal antibody; immunoperoxidase and hematoxylin counterstain; ×400). (C) HCL cells express phospho-ERK (red) in the cytoplasm and the nucleus (anti–alkaline phosphatase immunostaining and hematoxylin counterstain; ×400).

Trimming of HCL projections by BRAF inhibition. Confocal immunofluorescence staining for phalloidin (green), annexin V/ANXA5 (red), and the nuclear dye Draq5 (blue) in HCL cells purified from the peripheral blood of a representative patient with HCL and treated in vitro with vehicle DMSO (Ctrl) or Drug (vemurafenib 1 µM) for 48 hours (363× optical magnification with oil immersion). Upon drug treatment, first the hairy projections (rich in filamentous actin and thus stained in green by phalloidin) almost completely disappear while the leukemic cells are still alive (ie, negative for annexin V; top 2 cells in the right panel); afterward, apoptosis ensues (bottom 4 cells in the right panel). This figure is reproduced from Falini et al.15 DMSO, dimethyl sulfoxide.
Trimming of HCL projections by BRAF inhibition. Confocal immunofluorescence staining for phalloidin (green), annexin V/ANXA5 (red), and the nuclear dye Draq5 (blue) in HCL cells purified from the peripheral blood of a representative patient with HCL and treated in vitro with vehicle DMSO (Ctrl) or Drug (vemurafenib 1 µM) for 48 hours (363× optical magnification with oil immersion). Upon drug treatment, first the hairy projections (rich in filamentous actin and thus stained in green by phalloidin) almost completely disappear while the leukemic cells are still alive (ie, negative for annexin V; top 2 cells in the right panel); afterward, apoptosis ensues (bottom 4 cells in the right panel). This figure is reproduced from Falini et al.15 DMSO, dimethyl sulfoxide.
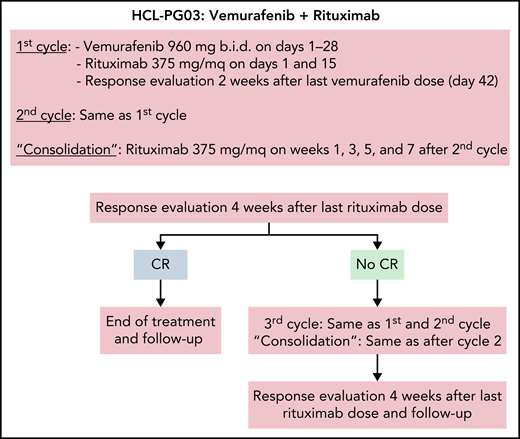
Treatment schema of the vemurafenib plus rituximab regimen. Schedule of vemurafenib plus rituximab used in the trial,35 where, however, in no patients a third cycle was delivered. The 2-week interval after vemurafenib dosing in each cycle was to allow enough time for adequate normal bone marrow (BM) recovery following clearance of the leukemic cell infiltration (especially after the first cycle) and, thus, for optimal reading of the BM biopsy scheduled after each cycle; however, outside a trial, interim BM biopsies are not typically needed, and the 2-week interval might be omitted from each cycle.
Treatment schema of the vemurafenib plus rituximab regimen. Schedule of vemurafenib plus rituximab used in the trial,35 where, however, in no patients a third cycle was delivered. The 2-week interval after vemurafenib dosing in each cycle was to allow enough time for adequate normal bone marrow (BM) recovery following clearance of the leukemic cell infiltration (especially after the first cycle) and, thus, for optimal reading of the BM biopsy scheduled after each cycle; however, outside a trial, interim BM biopsies are not typically needed, and the 2-week interval might be omitted from each cycle.
In a subsequent investigator-driven phase-2 trial, patients with relapsed/refractory HCL and a median of 3 prior therapies (including 37% refractory to PAs) were treated by adding rituximab (375 mg/m2, 8 doses over 18 weeks) to vemurafenib (960 mg twice daily for a total of 8 weeks, starting with rituximab) (Figure 3). The rationale was to eradicate vemurafenib-resistant HCL cells with the anti-CD20 monoclonal antibody.35 Addition of rituximab to vemurafenib increased CRs from 35% (of historical vemurafenib monotherapy33) to ≥87%.35 Moreover, the CR was achieved more quickly (4 vs 8 weeks), thus enabling to reduce median treatment duration from 16-18 weeks33 to 8 weeks.35 Importantly, 65% of patients achieving CR were MRD− (vs none with vemurafenib monotherapy33), and survival free from relapse of cytopenias (RFS) was 85% at 34 months of median follow-up after stopping treatment (vs a median RFS of 9 months after vemurafenib alone33). These results compare favorably not only with Moxe but also with PA plus rituximab, which in patients with a median of 3 prior therapies (none of whom having disease refractory to PA) led to an 88% CR rate and to an 87% RFS at a median follow-up of 78 months (Table 1). Vemurafenib plus rituximab was also effective in patients relapsed after Moxe.39
BRAF inhibitors, although not yet approved for HCL, are often used off-label in routine practice due to their widespread availability as on-label treatment of various BRAF-mutated solid cancers. Here, we present 3 challenging cases with refractory/relapsed HCL, who were treated with BRAF inhibitors alone or in combination with rituximab, to provide criteria for the diagnosis and therapeutic management of this difficult setting of patients.
Diagnosis of refractory/relapsed HCL
Outside referral centers, a challenging issue may be differentiating HCL from HCL-variant. Thus, in a disease like HCL that is usually well controlled by PAs, pathology revision should be advised, particularly in case of no response or early relapse. Response to cladribine should be diagnosed in a BM biopsy performed not earlier than 4 to 6 months after treatment because the drug antileukemic effect can be delayed.8 Furthermore, in the absence of cytopenias, persistence of minimal, or even morphologically appreciable, residual HCL disease does not typically require treatment.8
Patients with refractory/relapsed HCL who are candidates for BRAF inhibitors should be investigated for the BRAF-V600E mutation by molecular techniques40-43 and/or immunohistochemical staining of BM paraffin sections with a specific mouse monoclonal antibody (VE1)44-46 (Figure 1B). We do not prefer any single technique in this regard, but recommend to avoid those with lower sensitivity (eg, Sanger sequencing) because they are not suited to detect the mutation when leukemic cells are few, as often occurs in peripheral blood samples and fibrotic BM aspirates of patients with HCL. BRAF-V600E was present in >95% patients with refractory/relapsed HCL we investigated, including those relapsed up to 30 years after initial diagnosis (Falini and Tiacci, personal observation, February 2021). This extraordinary stability over time further confirms that BRAF-V600E is the genetic hallmark of HCL.
Dissemination of HCL to sites other than blood, marrow, and spleen (eg, lymph node or skin) is rare47 and usually occurs after multiple relapses (Figure 4A). Under these circumstances, only a paraffin-embedded biopsy sample is often available for diagnosis. Leukemic cells usually retain the typical HCL morphology (Figure 4B) and immunophenotype (eg, expression of B-cell antigens [Figure 4C] and ANXA13,4,48-50 [Figure 4D]). Expression of BRAF-V600E can be demonstrated by IHC,44-46 and its retention even in unusual sites further supports its clonal stability during the course of the disease.

Extramedullary involvement in refractory/relapsed HCL. (A) Computed tomography scan shows a large abdominal mass (asterisk), marked splenomegaly, and enlarged mediastinal lymph nodes (not shown). (B) Needle biopsy of the abdominal mass reveals infiltration of pancreatic glands by morphologically typical HCL cells with wide clear cytoplasm (hematoxylin-eosin; ×400). (C) HCL cells (red) strongly express the CD19 antigen (APAAP immunostaining, hematoxylin counterstaining; ×400). (D) HCL cells (brown) are also annexin-A1+ (immunoperoxidase staining; hematoxylin counterstaining; ×400). APAAP, alkaline phosphatase anti-alkaline phosphatase.
Extramedullary involvement in refractory/relapsed HCL. (A) Computed tomography scan shows a large abdominal mass (asterisk), marked splenomegaly, and enlarged mediastinal lymph nodes (not shown). (B) Needle biopsy of the abdominal mass reveals infiltration of pancreatic glands by morphologically typical HCL cells with wide clear cytoplasm (hematoxylin-eosin; ×400). (C) HCL cells (red) strongly express the CD19 antigen (APAAP immunostaining, hematoxylin counterstaining; ×400). (D) HCL cells (brown) are also annexin-A1+ (immunoperoxidase staining; hematoxylin counterstaining; ×400). APAAP, alkaline phosphatase anti-alkaline phosphatase.
Finally, as shown in case 2, other pathological conditions that may confound the cause of cytopenias or the interpretation of response to therapy, including therapy-related myelodysplasia/ acute myeloid leukemia (AML) or iatrogenic BM hypoplasia, should be excluded.
Case 1: HCL patient with previous splenectomy and multiple relapses
This 68-year-old male received many treatments over the previous 3 decades, leading to remissions followed by relapses and including splenectomy, interferon (repeated several times), pentostatin (repeated twice), rituximab, and cladribine (repeated 3 times). The patient presented again in 2013 with white blood cell (WBC) 9 × 109/L (10% neutrophils), hemoglobin (Hb) 9.2 g/dL, and platelets 146 × 109/L. The BM biopsy was heavily infiltrated by HCL. BRAF-V600E was detected in the peripheral blood.
Therapeutic approach What should have been the best therapeutic option for this patient in 2013? Adding rituximab to PAs was excluded because both these agents had been already administered and resulted in progressively shorter-duration responses or no response (as was the case for both rituximab and the last course of cladribine). Moreover, PAs are myelotoxic, especially in patients with scarce BM reserve. The same applies to bendamustine plus rituximab whose activity in refractory/relapsed HCL (ORR 100%; 50% to 67% durable CRs, mostly MRD−) was subsequently reported in a small number of patients.51 Chemotherapy-free treatment with Moxe11,13,14,52 was not an option in 2013 and would have been anyway suboptimal for our splenectomized patient because it is less effective in patients with previous splenectomy or marked splenomegaly.19
Therefore, he was enrolled in our HCL-PG01 study.33 The patient received vemurafenib monotherapy for 14 weeks with rapid resolution of cytopenias and reduction of BM leukemic infiltration. He experienced edema and cutaneous rash of grade 2 and acute pancreatitis of grade 3 that promptly resolved after stopping vemurafenib (960 mg twice daily) and did not recur upon completing therapy at lower doses (480 mg twice daily for 2 weeks followed by 720 mg twice daily for 10 weeks). Although hematological response after stopping therapy lasted ∼7 months (as neutrophils fell slightly below the 1.5 × 109/L threshold defining relapse in HCL), he required further treatment ∼10 months later (because of neutrophils < 1.0 × 109/L and mild anemia). Vemurafenib (720 mg twice daily) was delivered for 12 weeks uneventfully, resulting only in a short-term hematological response. Then he was started on maintenance with low-dose vemurafenib (240 mg twice daily, alternating 4 weeks on- with 4 weeks off-therapy) for ∼1 year, without further progression of cytopenias. However, ∼6 months after stopping maintenance, he became severely pancytopenic and, after a brief unsuccessful course of interferon, he was enrolled at age of 72 years in our HCL PG-03 phase-2 trial of vemurafenib (8 weeks) plus rituximab (8 doses).35
Interestingly, despite previous suboptimal responses to vemurafenib and refractoriness to rituximab, he achieved a MRD− CR, after only 8 weeks of vemurafenib plus 2 rituximab doses. MRD negativity by allele-specific polymerase chain reaction in BM aspirate was still maintained at the last follow-up ∼20 months after the end of treatment. He experienced only an infusion reaction due to rituximab and transient fatigue, dyspepsia, warts, and liver function test abnormalities due to vemurafenib (given mostly at 960 mg twice daily; 92% relative dose intensity).
Case 2: persistent cytopenias after vemurafenib therapy for refractory HCL
This 67-year-old male also received many treatments over the previous 3 decades, leading to remissions followed by relapses and including interferon (repeated 4 times), cladribine (repeated 3 times), and rituximab. The patient presented again in 2015 with marked splenomegaly (19 cm as longest spleen diameter on echography) and severe pancytopenia: WBC 0.66 × 109/L (29% neutrophils), Hb 8.4 g/dl, and platelets 18.0 × 109/L. He was transfusion-dependent for red blood cells and platelets. The BM was hypercellular and markedly infiltrated by HCL cells (70%). BRAF-V600E was present, and he was enrolled in our HCL-PG03 trial. He received 4 weeks of vemurafenib at 960 mg twice daily and 2 doses of rituximab on days 1 and 15, which were complicated by only mild arthralgias, warts, asymptomatic abnormalities of liver function tests, and a grade-2 infusion-related reaction. However, the patient remained severely pancytopenic and transfusion-dependent, which was unexpected for this regimen usually leading to rapid improvement of blood counts.35 Therefore, a BM evaluation was performed.
Therapeutic approach The critical issue here is to establish whether pancytopenia was related to lack of response to treatment (persistent BM infiltration by HCL), to scarce BM reserve, or to a therapy-related myeloid neoplasm from previous chemotherapies.
As compared with the pretherapy BM biopsy (Figure 5, upper left), the posttreatment one showed a complete disappearance of HCL (Figure 5, upper right), with MRD negativity molecularly documented also in the BM aspirate. However, the BM appeared myelodysplastic with monolobated megakaryocytes and about 10% to 15% CD34+ cells (Figure 5, bottom panels). Myelodysplasia (MDS), including increased CD34+ cell count, was retrospectively observed in the BM sample taken before starting treatment, where it had been overlooked due to overwhelming HCL infiltration (Figure 5, upper left). BM cytogenetic analysis showed monosomy 7 in 75% of metaphases, pointing to a therapy-related MDS.53 He was started on anti-MDS/AML treatment (first decitabine, then intensive chemotherapy) but died of progressive disease 6 months later.

Persistent pancytopenia after therapy with vemurafenib plus rituximab for HCL relapsed following multiple lines of chemotherapy. Upper left panel, immunohistochemical staining of BM biopsy with an anti-CD19 monoclonal antibody before starting vemurafenib plus rituximab shows considerable infiltration by CD19+ leukemic hairy cells (brown). (Leica immunostaining; immunoperoxidase; hematoxylin counterstaining; ×400). Upper right panel, BM biopsy after vemurafenib plus rituximab therapy showing complete disappearance of HCL. The thin arrow indicates a single residual normal CD79a+ plasma cell (brown). The thick arrow points to a monolobated megakaryocyte (Leica immunostaining; immunoperoxidase; hematoxylin counterstaining; ×400). Bottom left panel, the BM biopsy after therapy with vemurafenib plus rituximab shows, on ematoxylin-eosin (EE) staining, a myelodysplastic marrow with many monolobated megakaryocytes (thick arrow). The bottom right panel shows many monolobated megakaryocytes (thick arrow) and a significant percentage of CD34+ blast cells (brown, thin arrow) (Leica immunostaining; immunoperoxidase; hematoxylin counterstaining; ×400). Retrospectively, monolobated megakaryocytes (thick arrow, upper left panel) and an increased number of CD34+ cells (inset in the upper left panel) were present also in the BM biopsy before vemurafenib plus rituximab therapy, but they had been overlooked because of the predominant HCL infiltration.
Persistent pancytopenia after therapy with vemurafenib plus rituximab for HCL relapsed following multiple lines of chemotherapy. Upper left panel, immunohistochemical staining of BM biopsy with an anti-CD19 monoclonal antibody before starting vemurafenib plus rituximab shows considerable infiltration by CD19+ leukemic hairy cells (brown). (Leica immunostaining; immunoperoxidase; hematoxylin counterstaining; ×400). Upper right panel, BM biopsy after vemurafenib plus rituximab therapy showing complete disappearance of HCL. The thin arrow indicates a single residual normal CD79a+ plasma cell (brown). The thick arrow points to a monolobated megakaryocyte (Leica immunostaining; immunoperoxidase; hematoxylin counterstaining; ×400). Bottom left panel, the BM biopsy after therapy with vemurafenib plus rituximab shows, on ematoxylin-eosin (EE) staining, a myelodysplastic marrow with many monolobated megakaryocytes (thick arrow). The bottom right panel shows many monolobated megakaryocytes (thick arrow) and a significant percentage of CD34+ blast cells (brown, thin arrow) (Leica immunostaining; immunoperoxidase; hematoxylin counterstaining; ×400). Retrospectively, monolobated megakaryocytes (thick arrow, upper left panel) and an increased number of CD34+ cells (inset in the upper left panel) were present also in the BM biopsy before vemurafenib plus rituximab therapy, but they had been overlooked because of the predominant HCL infiltration.
Thus, cytopenias in a patient with HCL previously treated with genotoxic chemotherapy should not automatically be ascribed to persisting/relapsing HCL. In particular, a therapy-related MDS/AML should always be excluded by appropriate morphological, immunophenotypical (including CD34 immunostaining), and cytogenetic investigations of BM samples before starting BRAF inhibitors. This is also important because such agents block ERK signaling in BRAF V600E-mutated cells while they paradoxically activate ERK signaling in preneoplastic or neoplastic BRAF wild-type cells with mutant or active RAS. As consequence of this paradoxical effect, patients continuously treated with BRAF inhibitors may develop secondary neoplasms, mostly cutaneous squamous-cell carcinomas and keratoachantomas of little malignant potential,33,54 which mandates a dermatological examination every month and a CT scan every 6 months during drug dosing. Although the risk of developing colonic polyps may also be increased in patients with melanoma taking BRAF inhibitors for >2 years,55 in HCL these drugs are usually given for a few months (Table 1), and endoscopy surveillance seems unnecessary. Moreover, in one patient with metastatic melanoma receiving vemurafenib, this BRAF inhibitor accelerated the growth of a NRAS-mutated chronic myelomonocytic leukemia, which had gone unrecognized despite a preexisting rising leukocytosis.56 This side effect of vemurafenib was managed by changing drug dosing from continuous to intermittent for allowing the leukocyte count to decrease during the drug-free intervals.56 Our patient’s myelodysplastic clone with monosomy 7 lacked RAS mutations but harbored mutations typical of 7q−/−7 myeloid neoplasms (ie, U2AF1 [Q157P], CUX1 [N1301Sfs], and PTPN11 [Q60R]).57-59 Because PTPN11 mutation promotes RAS signaling,60 vemurafenib could have transiently favored MDS clone growth during the few weeks of drug dosing. However, the fact that over the following months this clone did not show any dependency on vemurafenib (as opposed to the chronic myelomonocytic leukemia clone described in56) rather suggests that it had an intrinsically malignant behavior driven by its therapy-related origin and its multiple genetic lesions known to portend poor prognosis in myeloid malignancies.53,58,59,61
Case 3: HCL patient with active uncontrolled infections
A 47-year-old man was diagnosed with HCL in 1990. He then received 3 courses of interferon for over 20 years, followed by a first course of weekly cladribine plus rituximab in 2011 and a second one in 2015, which was prematurely discontinued due to febrile neutropenia and resulted in a PR. Upon relapse in 2016, he resumed interferon with temporary benefit but then presented to us severely pancytopenic (WBC 3.4 × 109/L, with 85% leukemic hairy cells and 0.041 neutrophils × 109/L, Hb 7.1 g/dL; platelets 10 × 109/L) and transfusion-dependent for red blood cells and platelets. A CT scan highlighted multiple bilateral lung consolidations pointing to pneumonia, without fever, cough, or dyspnea. Blood tested negative for galactomannan. Broad-spectrum empirical antibacterial therapy was instituted.
Therapeutic approach Because of disease-related neutropenia and monocytopenia,62 as well as profound neutropenia and T-cell depletion secondary to therapy with PAs,63,64 approximately 25% of patients with HCL experience a bacterial or opportunistic65-68 infection at the onset or during the course of the disease.
Patients with HCL with mild neutropenia and non–life-threatening infection should receive broad-spectrum antibiotics. Antileukemic therapy should be delayed until infection control or resolution. Cases presenting with severe pancytopenia and sepsis or serious opportunistic infections should be immediately treated with antimicrobial drugs and, ideally, with antileukemic therapy and growth factors to accelerate neutrophil recovery. However, PAs should be avoided in this setting because they induce neutropenia and marked T-cell depletion. Because of the risk of hemolytic uremic or capillary leak syndrome,69 Moxe may be disfavored, especially if the patient has (or is at risk for) sepsis-related hemodynamic decompensation or respiratory failure. The off-label use of BRAF inhibitors with or without rituximab likely represents the best therapeutic option for these patients.70-73 We prefer to administer vemurafenib in combination with rituximab because this regimen has a quicker and deeper effect than vemurafenib alone.33,35 Moreover, the expected B-cell depletion from rituximab may not be so concerning because opportunistic infections in HCL are typically due to neutrophil, T-cell, or monocyte deficiencies.
Thus, the patient, 2 days after a bronchoalveolar lavage, was enrolled in our trial of vemurafenib plus rituximab.35 On days 2 and 3 of antileukemic treatment, he developed fever with hypotension. Pseudomonas aeruginosa grew from a blood culture, which prompted additional antibacterial therapy. On day 3, the bronchoalveolar lavage was galactomannan+, with Aspergillus terreus growing in culture. Hence, voriconazole was started, whereas vemurafenib was decreased from 960 to 480 mg twice daily, being catabolized by cytochrome P3A4 (strongly inhibited by voriconazole in vitro). Neutrophils increased to >1.5 × 109/L in 5 weeks, which contributed to resolve the severe infections. In the meantime, voriconazole was replaced with isavuconazole (a weaker inhibitor of CYP3A4), and vemurafenib was gradually reescalated to 960 mg twice daily. Side effects were a wart from vemurafenib and transient neutropenia from rituximab. He achieved CR (MRD+ in the BM) and relapsed with cytopenias ∼20 months later. Notably, among the 26 complete responders in our trial, all 4 cases who relapsed (at a median follow-up of almost 3 years) were MRD+ at the end of treatment,35 and 2 of them (including this patient) had received the lowest relative dose intensities of vemurafenib (67% in this case).
This case also offers the opportunity to address the question of how to manage a patient with HCL relapse after vemurafenib plus rituximab. Another course of vemurafenib plus rituximab can be effective.39 However, our patient had experienced only a relatively short relapse-free interval after this regimen. Furthermore, his disease had responded suboptimally to chemo-immunotherapy with some toxicity. Thus, Moxe would have been an attractive choice due to its targeted mechanism of action different from PAs, BRAF inhibitors, and rituximab, but it is authorized only in the United States. Thus, additional therapeutic options need to be considered for this setting of HCL relapse outside the United States.
The activity and safety of ibrutinib (for an indefinite duration mostly at 420 mg daily) was recently evaluated in 28 patients with refractory/relapsed HCL74 (median of 4 prior therapies), who had an unusually high frequency of apparently BRAF wild-type disease (7/27 evaluable cases, 26%). OR and CR rates were respectively 27% and 4% at 32 weeks, increasing to 48% and 15% at 48 weeks, and to 58% and 23% at any time, with rare MRD negativity. Recovery of neutrophils >1 × 109/L and platelets >10 × 109/L required a median of 8 and 20 weeks, respectively, vs ≤4 weeks for Moxe and for vemurafenib plus or minus rituximab. Thus, responses appear less frequent, less profound, and less rapid as compared to Moxe and to vemurafenib plus or minus rituximab (Table 1). At 3.5 years of median follow-up, 21% of patients with HCL had progressed on treatment, 28% had discontinued ibrutinib while not in remission (mainly due to adverse events), and 8% died on treatment (due to pneumonia). Hence, patients’ progression-free on-treatment or in-remission off-treatment were 43%. Unlike CLL, there was no mobilization of leukemic cells in the blood.74 Ibrutinib-related adverse reactions were mostly grade 1 to 2 and similar to those observed in other B-cell malignancies (diarrhea, fatigue, myalgia, nausea, bruising, infections, atrial fibrillation), but in HCL, there was also an appreciable rate of grade 3 to 4 toxicities, including 22% neutropenia, 22% thrombocytopenia, 16% lung infection, and 11% febrile neutropenia. Thus, ibrutinib may be particularly suited to patients with HCL having mild cytopenias and little risk of infections who do not require a quick recovery of blood counts. However, a general question to ask in the treatment of HCL, a disease associated with long survival even in relapsed/refractory cases, is whether patients are better served with short treatments affording relatively long intervals off any drugs compared with continuous drug dosing strategies, even assuming similar progression-free survival and safety of these 2 alternative approaches.
Another appealing rationale approach is to co-inhibit BRAF and its downstream target MEK for preventing the resistance that can develop during single BRAF inhibition in HCL and other BRAF-mutated tumors through MEK reactivation by alternative ways that bypass BRAF.33,75,76 Mechanisms of resistance to BRAF inhibitors are understudied in HCL due to absence of true HCL cell lines5,77,78 and the problem of obtaining sufficient numbers of purified HCL cells from patients, who usually have leukopenia and a difficult-to-aspirate marrow. In metastatic melanoma patients treated continuously with BRAF inhibitors until progression, several genetic mechanisms of acquired resistance (eg, RAS or MEK mutations) have been described that reactivate MEK.76 Conversely, in HCL, where BRAF inhibitors are usually delivered for a fixed and relatively short duration,33-35 genetic mechanisms of acquired resistance were reported only in 2 patients. One case had obtained an objective response to vemurafenib (delivered for 6 months) but relapsed 2 months after stopping the drug with a disease that had newly acquired 2 subclonal KRAS mutations and was refractory to vemurafenib retreatment.33 A second case received low-dose vemurafenib continuously for almost 6 years but, after a long remission, progressed due to several subclonal mutations of KRAS (n = 7) and MAP2K1/MEK1 (n = 2).79 These mutations independently and gradually developed over the years in a convergent evolution to reactivate MEK, such that adding the MEK inhibitor cobimetinib to vemurafenib led the patient in remission again.79 Conversely, we observed that, in about 50% of evaluable trial patients treated with vemurafenib for a median of only 16 weeks, residual HCL cells persisting in the BM biopsy at the end of drug dosing expressed phosphorylated ERK (the downstream target of MEK) despite being exposed to vemurafenib.33 This finding suggests a different mechanism of relatively quick, adaptive resistance that may be of nongenetic origin as also observed in melanoma.80,81 This mechanism may be rather mediated by the BM microenvironment, which in vitro protects HCL cells from vemurafenib-induced MEK/ERK dephosphorylation and apoptosis.22
The strategy of dual BRAF and MEK blockade was explored in a phase-2 American multicenter trial of the BRAF inhibitor dabrafenib plus the MEK inhibitor trametinib given indefinitely to patients with relapsed/refractory HCL. Preliminary results in 43 patients treated for a median of 17 months showed rates of OR, CR, and MRD-negativity of 78%, 49%, and 15%, respectively,82 with all responses ongoing at the data cutoff and half of them lasting ≥18 months (Table 1). The most frequent adverse events were pyrexia, chills, hyperglicemia, nausea, peripheral edema, cough, and fatigue; 49% of patients experienced grade 3 to 4 adverse events, and 12% discontinued treatment after an adverse event. Overall, although toxicity does not appear minimal, the depth of response to continuous dabrafenib plus trametinib dosing seems higher than that of fixed-duration vemurafenib or dabrafenib, but the latter strategy can be effectively repeated at relapse32-34 and does not keep the leukemic clone under the constant selection pressure to develop drug resistance.79
Coming back to our patient who relapsed after vemurafenib plus rituximab, he was enrolled, and is currently being treated, in our ongoing phase-2 HCL-PG04 trial (EudraCT 2017-001836-20), testing the sequential combination, in a step-wise manner, of fixed-duration treatments with the anti-CD20 monoclonal antibody obinutuzumab, the BRAF inhibitor vemurafenib, and the MEK inhibitor cobimetinib.
Open questions
The standard doses of vemurafenib (960 mg twice daily) and dabrafenib (150 mg twice daily) that are approved in BRAF-mutated solid tumors were used in prospective clinical trials of patients with relapsed/refractory HCL for a median of 8 to 18 weeks.33-35 Retrospective case series examining off-trial use of lower doses for a median of 3 to 3.8 months, while initially suggesting a high efficacy of alternative regimens mostly at 240 mg twice daily,31 later found that doses of vemurafenib <480 mg twice daily and of dabrafenib <150 mg twice daily were actually associated with shorter treatment-free survival.32 In the trial of rituximab plus vemurafenib 960 mg twice daily, receipt of a relative vemurafenib dose intensity <70% seemed to be associated with treatment failure.35 In the absence of ad hoc prospective clinical trials formally comparing the efficacy and safety of standard vs lower BRAF inhibitor doses, we use the standard dose as almost half of patients tolerate it without needing dose reductions, whereas most of the remaining patients temporarily require mild dose reductions and are then able to reescalate the drug dose.33-35
As to the duration of treatment with BRAF inhibitors, we favor to shorten it as much as possible by adding rituximab to 8 weeks of vemurafenib,35 which increases efficacy and decreases toxicity compared with monotherapy with vemurafenib for a longer duration.31 If a BRAF inhibitor must be delivered alone, for the reasons explained in case 3, we disfavor indefinite dosing until progression and rather favor a fixed duration of ∼3 to 4 months (consistent with clinical trials33-35), followed by BM biopsy to differentiate between CR and PR so as to tailor the frequency of subsequent blood cell count monitoring.
Regarding which BRAF inhibitor to choose, among the 3 that are approved and used in BRAF-mutated melanoma (without major differences in efficacy) (ie, vemurafenib, dabrafenib, and encorafenib), most of the experience in HCL has been gained with vemurafenib, which has also been tested in combination with rituximab, but dabrafenib seems similarly effective and has been successfully used in patients relapsing after vemurafenib.32-35
Another open question is how to treat patients with HCL during the COVID-19 pandemic, especially until mass vaccination will have reduced severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) circulation in the population to a minimal level. If antileukemic therapy is urgently needed, and the patient has not been vaccinated against SARS-CoV2, a BRAF inhibitor is a safe and effective option as it avoids the myelotoxicity and immune-suppression of chemotherapy that can worsen COVID-19 severity,83 whereas rituximab should be delayed until the patient has completed the vaccination program and added to vemurafenib only if the patient does not respond to the latter drug. Considering that anti–SARS-CoV2-2 messenger RNA vaccination elicits a potent T-cell–mediated response84 that might still confer some protection from severe COVID-19 even in the absence of humoral immunity to the virus,85 treatment with a BRAF inhibitor, potentially even in combination with rituximab, could be considered as an alternative to chemotherapy with PA also in HCL patients lacking a robust serological response to anti-COVID-19 vaccination, a scenario that in chronic lymphocytic leukemia is frequent in treatment-naive patients and is constant in those with recent (<12 months) rituximab exposure,86 and that could worsen the course of a SARS-CoV2 infection developing after T-cell depleting chemotherapy with PA.
The role of additional drugs effective in other chronic mature B-cell malignancies, such as venetoclax87 and obinutuzumab, should be investigated in refractory/relapsed HCL. The same applies to anti-CD19 chimeric antigen receptor T cells, although low levels of autologous T cells due to previous PAs treatment may represent a potential obstacle to this approach.
Acknowledgments
The work of the authors has been supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC, Metastasis/5-per-mille grant no. 21198 to B.F., Investigator Grant 14447 to E.T.), the Hairy Cell Leukemia Foundation (annual grants from 2011 to 2016 to B.F. and E.T.), the Fondation ARC pour la Rechèrche sur le Cancer (2015 Leopold Griffuel Prize to B.F.), the Ministero della Salute (grant RF-2016-02362264 to E.T.), the European Research Council (ERC-FP7-2013 Consolidator Grant 617471 to E.T.), and the Leukemia and Lymphoma Society (Scholarship in Clinical Research 2030-14 to E.T.).
Authorship
Contribution: B.F. and E.T. designed and wrote the paper; L.D.C. managed the patients and provided related clinical information; and all authors read and approved the final version of the manuscript.
Conflict-of-interest disclosure: B.F. received research funding from Roche and holds a patent on the use of mutant BRAF as HCL biomarker. E.T. was a consultant for Innate Pharma, received research funding from Roche and travel cost reimbursement from Abbvie and Shire, and holds a patent on the use of mutant BRAF as HCL biomarker. L.D.C. declares no competing financial interests.
Correspondence: Brunangelo Falini, Section of Hematology and Center for Hemato-Oncological Research (CREO), Department of Medicine and Surgery, University of Perugia and Hospital Santa Maria della Misericordia, Piazzale Menghini 8, 06132 Perugia, Italy; e-mail: brunangelo.falini@unipg.it; and Enrico Tiacci, Section of Hematology and Center for Hemato-Oncological Research (CREO), Department of Medicine and Surgery, University of Perugia and Hospital Santa Maria della Misericordia, Piazzale Menghini 8, 06132 Perugia, Italy; e-mail: enrico.tiacci@unipg.it.
Requests for data sharing may be submitted to Brunangelo Falini and Enrico Tiacci (brunangelo.falini@unipg.it and enrico.tiacci@unipg.it).
