

TO THE EDITOR:
Pulmonary arterial hypertension (PAH) is a clinical concern in patients with β-thalassemia because of the associated risks of right-sided heart failure and death.1 Several risk factors have been proposed to increase the risk of PAH in this patient population, including chronic anemia, hemolysis, iron overload, vasculopathy, and hypocoagulability, yet management options remain limited.1,2 Prevalence rates exceeding 50% have been historically reported using various echocardiography-based cutoffs, with higher rates observed in non–transfusion-dependent, splenectomized, and older adults.2 Approximately 10 years ago, we conducted a large, multicenter study with a dedicated protocol for right heart catheterization (RHC) in patients with β-thalassemia with echocardiography values suggestive of PAH and confirmed a true prevalence rate of 2.1%.3 In this study, we provide long-term data on mortality in this subgroup of patients with confirmed PAH on RHC.
This was a long-term follow-up of patients previously recruited in a multicenter, cross-sectional study of patients with β-thalassemia followed at 8 comprehensive care centers taking part in the Italian Webthal project. (This trial was registered at www.clinicaltrials.gov as #NCT01496963.)3 Institutional review boards at participating centers approved the study protocol, and all participants signed a written informed consent before inclusion in the original study. Details of the original study have been previously described.3 In brief, adults (≥18 years) with a diagnosis of β-thalassemia major or intermedia and without chronic restrictive lung disease or a left ventricular ejection fraction ≤50% (n = 1309) were recruited between January 2012 and January 2013. Patients then underwent screening transthoracic echocardiography using continuous-wave Doppler sampling of the peak tricuspid-valve regurgitant jet velocity (TRV) to calculate the systolic pulmonary artery pressure (sPAP) and were divided into 3 groups4: pulmonary hypertension (PH) unlikely (n = 1234), sPAP ≤36 mm Hg or TRV ≤3.0 m/s; PH possible (n = 28), sPAP >36 and <40 mm Hg or TRV >3.0 and <3.2 m/s; and PH likely (n = 47), sPAP ≥40 mm Hg or TRV ≥3.2 m/s. After excluding patients with chronic cardiopulmonary disease and those unfit for an invasive procedure, 33 of the 47 patients with PH likely underwent RHC. Among those patients, 31 had PH with a mean PAP of ≥25 mm Hg, and PAH was confirmed in 27 patients with a pulmonary capillary wedge pressure ≤15 mm Hg (precapillary PH).4
For this study, we followed 24 patients with confirmed PAH on RHC until March 2021, death, or loss to follow-up. Three patients had transitioned care to other institutions immediately after the original study and were not included in this analysis. For each patient, we retrieved data at PAH diagnosis (baseline) for demographics (age and sex), splenectomy status, hemoglobin and serum ferritin levels (mean over previous 10 years), and functionality using the New York Heart Association classification and 6-minute walk test. We also retrieved baseline echocardiography (left ventricular ejection fraction, TRV, sPAP, tricuspid annular plane systolic excursion, and right atrium area) and RHC (mean PAP, sPAP, cardiac index, pulmonary vascular resistance [PVR], and vasoreactivity5) values. Information regarding use of PAH-related therapies and patients’ hemoglobin, serum ferritin, and echocardiography values at the last observation was retrieved.
Descriptive statistics are presented as median and interquartile range (IQR) or percentages. Comparisons were made using the Mann-Whitney U or Kruskal–Wallis tests for continuous variables and the Fisher’s exact test for categorical variables. Kaplan-Meier survival curves were constructed to estimate cumulative survival, and the log-rank test was used for comparisons of survival curves. Receiver operating characteristic curve analysis was used to estimate area under the curve for predictive variables. All P values were 2-sided, with the level of significance set at <.05.
A total of 24 patients (50% male) with a median age of 46.5 years (IQR: 39.3-59) were included in this analysis. The median follow-up time was 4 years (IQR: 1-6, minimum: 0.5, maximum: 9). Thirteen patients died during the observation period, giving a crude all-cause mortality rate of 54.2% (95% confidence interval [CI], 32.9-74.5). Three patients died due to hepatic disease or sepsis, whereas in 10 patients, death was attributed to PAH, giving a crude PAH-related mortality rate of 41.7% (95% CI, 22.1-63.4). The cause of death was right-sided heart failure in 9 patients and pulmonary embolism in 1 patient. The Kaplan-Meier survival curve for PAH-related mortality is illustrated in Figure 1A. The median survival time was 9 years. Cumulative PAH-related mortality-free survival estimates at 1, 2, and 5 years were 78%, 65%, and 60%, respectively.
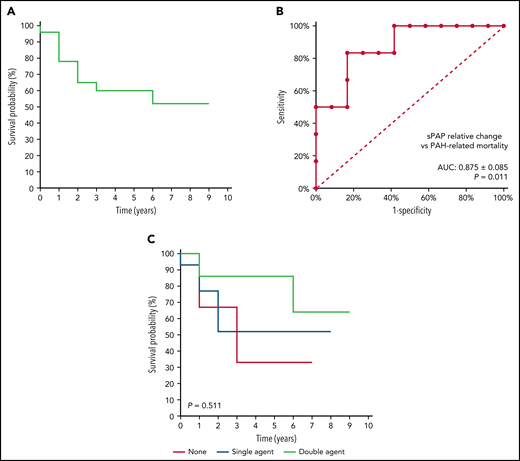
PAH-related mortality in patients with confirmed PAH. (A) Kaplan-Meier survival curve for PAH-related mortality. (B) Receiver operating characteristic curve for relative change in sPAP as a predictor of PAH-related mortality. (C) Kaplan-Meier survival curve for PAH-related mortality by receipt of PAH-related therapy. AUC, area under the curve.
PAH-related mortality in patients with confirmed PAH. (A) Kaplan-Meier survival curve for PAH-related mortality. (B) Receiver operating characteristic curve for relative change in sPAP as a predictor of PAH-related mortality. (C) Kaplan-Meier survival curve for PAH-related mortality by receipt of PAH-related therapy. AUC, area under the curve.
Comparisons of baseline parameters in patients who died due to PAH and those who did not are summarized in Table 1. There were no statically significant differences in demographics, thalassemia diagnosis, splenectomy status, hemoglobin and serum ferritin levels, and functional status between patients who died due to PAH and those who did not, noting that all patients who died were splenectomized and most had β-thalassemia intermedia. Baseline echocardiography and RHC values were also statistically comparable, with lower sPAP and cardiac index and higher PVR and vasoreactivity in patients who died due to PAH than those who did not (Table 1).
Comparison of study parameters in patients who died due to pulmonary arterial hypertension and those who did not
| Parameter | All (n = 24) | PAH-related mortality | P | |
|---|---|---|---|---|
| Yes (n = 10) | No (n = 14) | |||
| Demographics | ||||
| Baseline age in years | 46.5 () | 44.5 () | 48 () | .709 |
| Male, % | 50 | 40 | 57.1 | .680 |
| Thalassemia diagnosis, % | ||||
| Thalassemia major | 41.7 | 40 | 42.9 | 1.000 |
| Thalassemia intermedia | 58.3 | 60 | 57.1 | |
| Splenectomized, % | 91.7 | 100 | 85.7 | .493 |
| Functional status (baseline) | ||||
| NYHA class, % | ||||
| I or II | 34.8 | 44.4 | 28.6 | .657 |
| III or IV | 65.2 | 55.6 | 71.4 | |
| 6-MWD in m | 480 () | 470 () | 486.5 () | .711 |
| Right heart catheterization (baseline) | ||||
| mPAP in mm Hg | 41.5 () | 45.5 () | 40.5 () | .437 |
| sPAP in mm Hg | 63 () | 60 () | 66 () | .563 |
| Cardiac index in l/min/ | 3.4 () | 2.9 () | 3.8 () | .877 |
| PVR in dyn-s- | 500.9 () | 611.9 () | 470.9 () | .267 |
| Vasoreactivity*, % | 26.7 | 42.9 | 12.5 | .282 |
| Laboratory | ||||
| Hemoglobin in g/dL | ||||
| Baseline | 9.2 () | 9.7 () | 9.2 () | .522 |
| Change from baseline | () | () | () | .414 |
| Serum ferritin in ng/mL | ||||
| Baseline | 694 () | 550 () | 985.9 () | .077 |
| Change from baseline | () | () | () | .710 |
| Echocardiography | ||||
| LVEF in % | ||||
| Baseline | 60 () | 60 () | 60 () | .926 |
| Change from baseline | 0 () | () | () | .711 |
| TRV in m/s | ||||
| Baseline | 3.8 () | 3.7 () | 4.0 () | .752 |
| Change from baseline | () | () | () | .400 |
| sPAP in mm Hg | ||||
| Baseline | 68.5 () | 65 () | 71 () | .931 |
| Change from baseline | () | () | () | .024 |
| TAPSE in cm | ||||
| Baseline | 20 () | 19.3 () | 22 () | .235 |
| Change from baseline | () | () | () | .661 |
| Right atrium area in | ||||
| Baseline | 31 () | 32 () | 31 () | .630 |
| Change from baseline | () | 0 () | () | 1.000 |
| Parameter | All (n = 24) | PAH-related mortality | P | |
|---|---|---|---|---|
| Yes (n = 10) | No (n = 14) | |||
| Demographics | ||||
| Baseline age in years | 46.5 () | 44.5 () | 48 () | .709 |
| Male, % | 50 | 40 | 57.1 | .680 |
| Thalassemia diagnosis, % | ||||
| Thalassemia major | 41.7 | 40 | 42.9 | 1.000 |
| Thalassemia intermedia | 58.3 | 60 | 57.1 | |
| Splenectomized, % | 91.7 | 100 | 85.7 | .493 |
| Functional status (baseline) | ||||
| NYHA class, % | ||||
| I or II | 34.8 | 44.4 | 28.6 | .657 |
| III or IV | 65.2 | 55.6 | 71.4 | |
| 6-MWD in m | 480 () | 470 () | 486.5 () | .711 |
| Right heart catheterization (baseline) | ||||
| mPAP in mm Hg | 41.5 () | 45.5 () | 40.5 () | .437 |
| sPAP in mm Hg | 63 () | 60 () | 66 () | .563 |
| Cardiac index in l/min/ | 3.4 () | 2.9 () | 3.8 () | .877 |
| PVR in dyn-s- | 500.9 () | 611.9 () | 470.9 () | .267 |
| Vasoreactivity*, % | 26.7 | 42.9 | 12.5 | .282 |
| Laboratory | ||||
| Hemoglobin in g/dL | ||||
| Baseline | 9.2 () | 9.7 () | 9.2 () | .522 |
| Change from baseline | () | () | () | .414 |
| Serum ferritin in ng/mL | ||||
| Baseline | 694 () | 550 () | 985.9 () | .077 |
| Change from baseline | () | () | () | .710 |
| Echocardiography | ||||
| LVEF in % | ||||
| Baseline | 60 () | 60 () | 60 () | .926 |
| Change from baseline | 0 () | () | () | .711 |
| TRV in m/s | ||||
| Baseline | 3.8 () | 3.7 () | 4.0 () | .752 |
| Change from baseline | () | () | () | .400 |
| sPAP in mm Hg | ||||
| Baseline | 68.5 () | 65 () | 71 () | .931 |
| Change from baseline | () | () | () | .024 |
| TAPSE in cm | ||||
| Baseline | 20 () | 19.3 () | 22 () | .235 |
| Change from baseline | () | () | () | .661 |
| Right atrium area in | ||||
| Baseline | 31 () | 32 () | 31 () | .630 |
| Change from baseline | () | 0 () | () | 1.000 |
All data presented as median (IQR) unless otherwise specified.
6-MWD, 6-min walk distance; LVEF, left-ventricular ejection fraction; mPAP, mean pulmonary arterial pressure; NYHA, New York Heart Association; TAPSE, tricuspid annular plane systolic excursion.
A positive acute response was defined as a reduction in mPAP of 10 mm Hg to reach an absolute value of 40 mm Hg with an increased or unchanged cardiac output.
Hemoglobin and serum ferritin levels only marginally changed during the period of observation (Table 1). Absolute changes in echocardiography values during the observation period are summarized in Table 1. There was a statistically significant difference in the median absolute change in sPAP between patients who died due to PAH and those who did not (+6.5 vs −21.6 mm Hg; P = .024). This value corresponded to a median relative change (absolute change/baseline × 100%) of +8.2% vs −31.6% (P = .010). On receiver operating characteristic curve analysis, the relative change in sPAP was a strong predictor of PAH-related mortality (area under the curve: 0.875 ± 0.085; P = .011) with a relative change of −25.6% having 100% sensitivity and a relative change of +12.0% having 100% specificity to predict PAH-related mortality (Figure 1B).
A total of 21 patients received PAH-related therapy after PAH diagnosis, with 14 (58.3%) receiving single-agent therapy and 7 (29.2%) receiving double-agent therapy. Therapies included bosentan, ambrisentan, sildenafil, tadalafil, macitentan, riociguat, and other angiotensin-converting enzyme inhibitors, calcium channel and β-blockers, and anticoagulants. Crude PAH-related mortality rate was 38.1% in patients receiving any PAH-related therapy. Crude PAH-related mortality rates were 66.7%, 42.9%, and 28.6% in patients receiving no, single-, and double-agent PAH-related therapy, respectively (P = .529). Cumulative PAH-related mortality-free survival estimates at 5 years were 33%, 52%, and 86%, respectively (log-rank χ2: 1.342; P = .511; Figure 1C). Compared with patients with no PAH-related therapy, patients with single- and double-agent therapy had a hazard ratio for death of 0.710 (95% CI, 0.143-3.52) and 0.352 (95% CI, 0.049-2.514), respectively. The median relative change in sPAP on echocardiography was higher in patients with double vs single agent vs no PAH-related therapy, but this did not reach statistical significance (−54.5% vs +1.5% vs −8.7%; P = .371).
Although limited by a small sample size, our study provided mortality estimates for patients with β-thalassemia with confirmed PAH and furthered our understanding of the detrimental impact of this morbidity, regardless of the underlying patient profile. A protective role for PAH and PAH-related mortality has been suggested for transfusion therapy in observational studies of non–transfusion-dependent β-thalassemia6,7 although PAH-related mortality was comparable in β-thalassemia major and intermedia in this cohort. Data on the benefit of pharmacologic therapy in patients at risk or with established disease are limited to a few small clinical trials.8,9 Although this study was not designed to evaluate the impact of therapy on PAH-related mortality, improvement in sPAP with the use of pharmacologic agents was associated with a lower mortality rate, especially for patients achieving >25% reduction from baseline. Randomized trials evaluating agents and combinations targeting PAH in this group of patients are needed. Larger longitudinal studies and disease-specific guidelines are also merited to establish best practices for routine screening and early intervention.
Authorship
Contribution: V.M.P., K.M.M., G.D., and G.L.F. conceived and designed the study; V.M.P., G.D., G.G., M.G., R.O., S.B., G.C., A. Pasanisi, F.L., M.C., R.M., P.M., I.T., A. Piga, M.D.C., B.G., and G.L.F. collected data; K.M.M. conducted statistical analysis; all authors reviewed and interpreted results; K.M.M. drafted the manuscript; all authors reviewed the manuscript for important intellectual content; all authors approved the manuscript before submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Gian Luca Forni, Center for Microcythemia, Congenital Anemia and Iron Dysmetabolism, Galliera Hospital, Via Volta 6, 16128 Genoa, Italy; e-mail: gianluca.forni@galliera.it.
Data are available upon request to the corresponding author.
There is a Blood Commentary on this article in this issue.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal