

Key Points
In telomere biology disorders, patients with autosomal dominant inheritance patterns, excluding TINF2, show the best OS.
The mode of inheritance should be incorporated into evidence-based clinical care for patients with telomere biology disorders.

Abstract
Dyskeratosis congenita related telomere biology disorders (DC/TBDs) are characterized by very short telomeres caused by germline pathogenic variants in telomere biology genes. Clinical presentations can affect all organs, and inheritance patterns include autosomal dominant (AD), autosomal recessive (AR), X-linked (XLR), or de novo. This study examined the associations between mode of inheritance with phenotypes and long-term clinical outcomes. Two hundred thirty-one individuals with DC/TBDs (144 male, 86.6% known genotype, median age at diagnosis 19.4 years [range 0 to 71.6]), enrolled in the National Cancer Institute’s Inherited Bone Marrow Failure Syndrome Study, underwent detailed clinical assessments and longitudinal follow-up (median follow-up 5.2 years [range 0 to 36.7]). Patients were grouped by inheritance pattern, considering AD-nonTINF2, AR/XLR, and TINF2 variants separately. Severe bone marrow failure (BMF), severe liver disease, and gastrointestinal telangiectasias were more prevalent in AR/XLR or TINF2 disease, whereas pulmonary fibrosis developed predominantly in adults with AD disease. After adjusting for age at DC/TBD diagnosis, we observed the highest cancer risk in AR/XLR individuals. At last follow-up, 42% of patients were deceased with a median overall survival (OS) of 52.8 years (95% confidence interval [CI] 45.5-57.6), and the hematopoietic cell or solid organ transplant-free median survival was 45.3 years (95% CI 37.4-52.1). Significantly better OS was present in AD vs AR/XLR/TINF2 disease (P < .01), while patients with AR/XLR and TINF2 disease had similar survival probabilities. This long-term study of the clinical manifestations of DC/TBDs creates a foundation for incorporating the mode of inheritance into evidence-based clinical care guidelines and risk stratification in patients with DC/TBDs. This trial was registered at www.clinicaltrials.gov as #NCT00027274.
Introduction
Telomeres, specialized nucleoprotein structures composed of (TTAGGG)n repeats at the ends of eukaryotic chromosomes, protect chromosome integrity during cell replication and maintain genome stability.1 Telomere biology disorders (TBD) include a spectrum of illnesses characterized by short to very short age-adjusted telomeres caused by defective telomere maintenance.
Dyskeratosis congenita (DC) was the first described and is the most well-known TBD.2-4 The classical DC phenotype consists of the mucocutaneous triad of dysplastic nails, oral leukoplakia, and lacy reticular skin pigmentation. The triad may be subtle or absent at presentation but progressive with time.5 Advances in understanding the pathophysiology of telomere-associated disease have led to the recognition of a spectrum of DC/TBD-related clinical features sometimes differentially affecting multiple organ systems and complicating the diagnosis of DC/TBDs.6 Some patients present in childhood with complex multisystem illnesses (eg, Hoyeraal-Hreidarsson syndrome [HH], Revesz syndrome [RS], or Coats plus syndrome [CS]), or bone marrow failure (BMF) and classic DC skin changes. Adults with TBDs may develop apparently isolated BMF, pulmonary fibrosis (PF), or liver disease (LD).7-11 Patients with DC/TBDs also have a significantly increased risk of cancer compared with the general population, especially head and neck squamous cell carcinoma (HNSCC).12 Additional severe complications include pulmonary arteriovenous malformations (PAVM), gastrointestinal bleeding, LD, and avascular osteonecrosis.6,13-15 The clinical spectrum of DC/TBDs may also include neurological, ophthalmic, dental, and immunologic abnormalities.6,16-19
Disease-causing variants have been reported in at least 15 genes regulating telomere maintenance (DKC1, TERC, TERT, NOP10, NHP2, ACD, TINF2, POT1, CTC1, STN1, WRAP53, RTEL1, PARN, NAF1, ZCCHC8).6,20 The underlying inheritance of these pathogenic variants is diverse. Depending on the gene affected, variants may be inherited in X-linked recessive (XLR), autosomal dominant (AD), or autosomal recessive (AR) patterns or occur de novo.6,20 The different inheritance patterns together with variable genetic penetrance, expressivity, and anticipation contribute to the phenotypic spectrum and may lead to variable disease manifestations within a family.6 Heterozygous germline variants in TERT, TERC, RTEL1, and PARN are more likely to present in adults with isolated disease, such as BMF or PF.7,8,21,22 Children with multisystem clinical presentations, such as HH or RS, are frequently reported to be carriers of XLR (DKC1), AR, and heterozygous TINF2 disease.23-26 Shortest telomeres have previously been witnessed in HH, RS, and DKC1- or TINF2-associated DC/TBDs.27
Most prior studies were cross-sectional or focused on organ-specific DC/TBD manifestations.5,11,13-16,28-30 Our study was based on the hypothesis that the genetic background underlying DC-related telomere biology disorders is associated with phenotype range and severity. We conducted a longitudinal comprehensive phenotype assessment across all organ systems in individuals with DC/TBDs to examine the association of gene and mode of inheritance with clinical manifestations and quantify the association of genotype subgroups with clinical outcomes and long-term survival.
Methods
Subjects
The National Cancer Institute’s (NCI) Institutional Review Board-approved longitudinal cohort study of inherited BMF syndromes (IBMFS) (clinicaltrials.gov NCT00027274, https://marrowfailure.cancer.gov) is a retrospective and prospective observational study.12,31 All participants or their legal guardians provide informed consent in accordance with Health and Human Services regulation 45 CFR 46. Detailed questionnaires are completed by participants, including personal medical and family history, and medical records are obtained and reviewed by medical experts. All participants enroll in the field cohort, and a subset of participants undergo detailed evaluation at the National Institutes of Health (NIH) Warren G. Magnuson Clinical Center, Bethesda, MD (clinic cohort).
Design
We included patients with TBDs, including DC, RS, HH, or CS enrolled in the NCI IBMFS study between January 1st 2002 and May 31st 2019. Medical records and questionnaires were collected and reviewed through September 15th 2020. Clinical diagnoses were confirmed by the presence of short (first through tenth percentile for age) or very short (<first percentile for age) telomeres in peripheral blood (PB) lymphocyte subsets by flow cytometry and fluorescent in situ hybridization (flow-FISH)32 and/or validated by DNA sequencing of DC-associated genes.
DC/TBD-related features may present late in adulthood, and short but not very short telomere length (TL) can occur in those with heterozygous variants (excluding TINF2).22,33,34 Therefore, we included family members of index cases with AR inheritance in RTEL1, TERT, ACD, and PARN if the relatives were heterozygous for potentially pathogenic variants with or without very short telomeres and/or DC triad features, PF, BMF, or LD (supplemental Table 1).
We included family members on whom the genotype could be inferred by mode of inheritance and reported clinical information (presence of very short telomeres, PF, BMF, and/or hematologic malignancies) (supplemental Table 2).35
Analysis
Medical data were collected starting from birth until death or last contact. Diagnoses were considered if they were (1) established at the time of NIH visit (clinic cohort) and/or (2) reported as established diagnoses by medical record review, self and/or family relation report (supplemental Table 3). A detailed phenotype assessment was done for 91 clinic cohort participants at the NIH Clinical Center, including subspecialist evaluations by hematologists, oncologists, otolaryngologists, dentists, dermatologists, psychiatrists, ophthalmologists, as well as clinical laboratory studies and medical imaging. Medical records and questionnaires were reviewed in detail for both the clinic (n = 91) and field cohorts (n = 140).
Germline genetic variants were deemed pathogenic based on clinical laboratory reports and/or by adaptation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) variant curation guidelines36 using the VarSeq v2.2.0 (Golden Helix, Inc., Bozeman, MT, http://www.goldenhelix.com)37 and VarSome38 software (https://varsome.com) (details in supplemental Tables 4 and 5).
We evaluated individuals based on their inheritance pattern: (1) AR and XLR were combined (AR/XLR); (2) TINF2-associated DC/TBD was considered separately because of the high occurrence of de novo variants (TINF2); and (3) AD refers to those with heterozygous variants, except for TINF2 disease (Figure 1B).5,11,16 TL was adjusted for age in our study through the use of normalized percentiles per age or z-scores.27
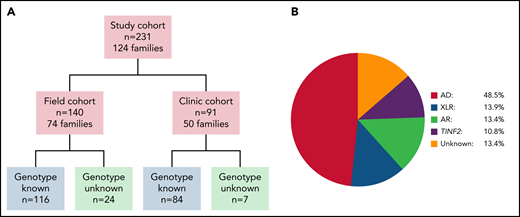
Overview of study cohort and distribution of inheritance pattern subgroups. (A) Cohort overview. Field cohort: participants completed questionnaires and the study team reviewed their medical records. 32 field cohort participants were also members of clinic cohort families. Clinic cohort: participants evaluated by specialists at the National Institutes of Health Warren G. Magnuson Clinical Center. (B) Schematic depiction of the distribution of inheritance pattern in the 231 study subjects. AD, autosomal dominant inheritance; AR, autosomal recessive inheritance; XLR, X-linked recessive inheritance; TINF2, TINF2 heterozygotes.
Overview of study cohort and distribution of inheritance pattern subgroups. (A) Cohort overview. Field cohort: participants completed questionnaires and the study team reviewed their medical records. 32 field cohort participants were also members of clinic cohort families. Clinic cohort: participants evaluated by specialists at the National Institutes of Health Warren G. Magnuson Clinical Center. (B) Schematic depiction of the distribution of inheritance pattern in the 231 study subjects. AD, autosomal dominant inheritance; AR, autosomal recessive inheritance; XLR, X-linked recessive inheritance; TINF2, TINF2 heterozygotes.
Severe BMF was defined as absolute neutrophil count (ANC) <500/mm3, platelet count <20 000/mm3, hemoglobin (Hb) <8.0 g/dL, or cytopenia requiring treatment (eg, transfusions or oral androgens). BMF was always considered severe if hematopoietic cell transplantation (HCT) had been performed and/or leukemia and/or myelodysplastic syndrome (MDS) had been diagnosed. Nonsevere BMF included ANC between 500 and <1500/mm3, platelet count between 20 000 and <150 000/mm3, and/or Hb ≥8g/dL less than normal for age. Diagnosis of MDS was considered if reported by medical or self-report. If available, diagnoses were verified (by original cytogenic and/or pathology report). LD included portal hypertension and/or liver cirrhosis/fibrosis (supplemental Table 3).
Mann-Whitney U test was used for continuous variables and Fisher’s exact test for categorical variables when comparing clinical features between subgroups. We used logistic regression models, calculated the odds ratios (ORs) and 95% confidence intervals (CIs) of clinical complications in relation to inheritance modes excluding individuals with unknown genotype. Associations between genotype and cancer and the influence of TL and genotype on survival were analyzed using multivariable Cox proportional hazards regression models. Proportional hazard assumption was tested by using Schoenfeld residuals; no significant violations were observed.
Survival probabilities for overall and transplant-free survival (TFS) were calculated using Kaplan Meier estimates; for comparison of survival probabilities, the log-rank test was used. TFS was defined as time from birth to death or any organ transplant (HCT, lung or liver transplantation). All statistical tests were 2-sided. P values <.05 were considered significant. Statistical analyses and image creation were performed using R (version 4.1.0, R Core Team [2021].39
Results
Participant characteristics
Two hundred thirty-one patients with DC/TBDs were enrolled, and 91 underwent comprehensive evaluations at the NIH (clinic cohort) (Table 1, Figure 1A). One hundred forty-four (62.3%) participants were male; when excluding participants with DKC1 variants, 56.3% of 199 individuals were male. Most patients self-reported as White (170/187 with data available). The median age at DC/TBD diagnosis was 19.4 years (range 0 to 71.6). The median age at last follow-up was 29.6 years (range 1.3 to 82.2), and the median duration of follow-up was 5.2 years (range 0 to 36.7) (Table 1).
Characteristics of study participants
| Entire cohort, n | Clinic cohort, n | Field cohort, n | |
|---|---|---|---|
| Number of families | 124 | 50 | 74* |
| Number of individuals | 231 | 91 | 140* |
| Male:female | 144:87 | 57:34 | 87:53 |
| Self-reported race/ethnicity | |||
| White | 170 | 83 | 87 |
| Asian | 6 | 2 | 4 |
| Black | 1 | 1 | 0 |
| Native American | 1 | 1 | 0 |
| Mixed | 9 | 3 | 6 |
| Unknown | 44 | 1 | 43 |
| Diagnosis categories | |||
| DC/TBD | 199 | 79 | 120 |
| HH | 23† | 10 | 13 |
| Revesz syndrome | 6‡ | 2 | 4 |
| Coats plus | 3§ | 0 | 3 |
| Median age at diagnosis in years (range)ǁ | 19.4 (0-71.6) | 22.3 (0-69.4) | 18.4 (0-71.6) |
| Patients diagnosed <18 y of age | 96 | 41 | 55 |
| Patients diagnosed ≥18 y of age | 111 | 50 | 61 |
| Median follow-up since diagnosis (range)ǁ,# | 5.2 (0-36.7) | 7.6 (0-30.2) | 2.7 (0-36.7) |
| Median age at follow-up in years (range) | 29.6 (1.3-82.2) | 29.2 (2.2-79.5) | 29.8 (1.3-82.2) |
| Number deceased at last follow-up | 97 | 32 | 65 |
| Gene known¶ | 200 | 84 | 116 |
| TERT (AR,AD) | 49 (2,47) | 14 (2,12) | 35 (0,35) |
| RTEL1 (AR,AD)# | 44 (15,29) | 24 (9,15) | 20 (6,14) |
| DKC1 (XLR) | 32 | 11 | 21 |
| TERC (AD) | 30 | 10 | 20 |
| TINF2 (AD) | 25 | 14 | 11 |
| PARN (AR,AD) | 8 (4,4) | 6 (3,3) | 2 (1,1) |
| CTC1 (AR) | 6 | 0 | 6 |
| WRAP53 (AR) | 3 | 2 | 1 |
| ACD (AR,AD) | 3 (1,2) | 3 (1,2) | 0 |
| Gene unknown | 31 | 7 | 24 |
| Inheritance pattern | |||
| AD | 112 | 42 | 70 |
| AR/XLR | 63 | 28 | 35 |
| TINF2 (de novo,AD) | 25 (13,12) | 14 (7,7) | 11 (6,5) |
| Entire cohort, n | Clinic cohort, n | Field cohort, n | |
|---|---|---|---|
| Number of families | 124 | 50 | 74* |
| Number of individuals | 231 | 91 | 140* |
| Male:female | 144:87 | 57:34 | 87:53 |
| Self-reported race/ethnicity | |||
| White | 170 | 83 | 87 |
| Asian | 6 | 2 | 4 |
| Black | 1 | 1 | 0 |
| Native American | 1 | 1 | 0 |
| Mixed | 9 | 3 | 6 |
| Unknown | 44 | 1 | 43 |
| Diagnosis categories | |||
| DC/TBD | 199 | 79 | 120 |
| HH | 23† | 10 | 13 |
| Revesz syndrome | 6‡ | 2 | 4 |
| Coats plus | 3§ | 0 | 3 |
| Median age at diagnosis in years (range)ǁ | 19.4 (0-71.6) | 22.3 (0-69.4) | 18.4 (0-71.6) |
| Patients diagnosed <18 y of age | 96 | 41 | 55 |
| Patients diagnosed ≥18 y of age | 111 | 50 | 61 |
| Median follow-up since diagnosis (range)ǁ,# | 5.2 (0-36.7) | 7.6 (0-30.2) | 2.7 (0-36.7) |
| Median age at follow-up in years (range) | 29.6 (1.3-82.2) | 29.2 (2.2-79.5) | 29.8 (1.3-82.2) |
| Number deceased at last follow-up | 97 | 32 | 65 |
| Gene known¶ | 200 | 84 | 116 |
| TERT (AR,AD) | 49 (2,47) | 14 (2,12) | 35 (0,35) |
| RTEL1 (AR,AD)# | 44 (15,29) | 24 (9,15) | 20 (6,14) |
| DKC1 (XLR) | 32 | 11 | 21 |
| TERC (AD) | 30 | 10 | 20 |
| TINF2 (AD) | 25 | 14 | 11 |
| PARN (AR,AD) | 8 (4,4) | 6 (3,3) | 2 (1,1) |
| CTC1 (AR) | 6 | 0 | 6 |
| WRAP53 (AR) | 3 | 2 | 1 |
| ACD (AR,AD) | 3 (1,2) | 3 (1,2) | 0 |
| Gene unknown | 31 | 7 | 24 |
| Inheritance pattern | |||
| AD | 112 | 42 | 70 |
| AR/XLR | 63 | 28 | 35 |
| TINF2 (de novo,AD) | 25 (13,12) | 14 (7,7) | 11 (6,5) |
Follow-up was defined as last contact with study center by treating physician, participant, or family through questionnaire, clinical report, e-mail, or phone call.
AD, autosomal dominant; ACD, ACD shelterin complex subunit and telomerase recruitment factor; AR, autosomal recessive; CTC1, CST telomere replication complex component 1; DC, dyskeratosis congenita; DKC1, dyskerin; HH, Hoyeraal-Hreidarsson syndrome; RTEL1, regulator of telomere elongation; TBD, telomere biology disorders; TERC, telomerase RNA component; TERT, telomerase reverse transcriptase; TINF2, TERF1 interacting nuclear factor 2; PARN, poly(A)-specific ribonuclease; WRAP53, WD repeat containing antisense to TP53; XLR, X-linked recessive.
32 field cohort participants were members of clinic cohort families.
2 AD, 18 AR/XLR, 3 unknown.
3 TINF2, 1 AR/XLR, 2 unknown.
3 AR/XLR.
The diagnosis was established postmortem for 24 patients: they were not included in the calculation of age at diagnosis or follow-up since diagnosis.
Includes 25 individuals with inferred genotype based on family history and inheritance pattern of genotype.
The follow-up period since diagnosis was significantly lower in the field vs the clinic cohort. There was a significant difference in the distribution of RTEL1 affected in the clinic vs field cohort.
The genetic cause of disease was identified in 200 (86.6%) participants (Table 1). Twenty-five individuals were heterozygous carriers of variants in RTEL1, TERT, ACD, or PARN where the index-case presented with biallelic disease. Pathogenic variants were confirmed in 176 individuals by clinical genetic testing. The genotype was inferred for 24 individuals based on the inheritance pattern (Figure 1B; supplemental Table 2). Pathogenic variants were most common in TERT, RTEL1, TERC, and DKC1. We performed the genotype-phenotype analyses by mode of inheritance, instead of only the affected gene, based on prior clinical observations that AR/XLR and TINF2 DC/TBDs tend to present in childhood.23,24,40-42 The predominant inheritance pattern was AD (n = 112, RTEL1, TERT, TERC, ACD, and PARN) (Figure 1B). There were 32 XLR and 31 AR individuals, with AR RTEL1 being most common (Table 1). Thirteen individuals had de novo TINF2 variants, and 12 had relatives heterozygous for the same TINF2 variant.
Flow FISH lymphocyte TLs were measured in 171 patients, and 133 had telomeres < first percentile for age. The prevalence of very short telomeres (< first percentile for age) was higher in AR/XLR and TINF2 compared with AD disease (supplemental Table 6; supplemental Figure 1). Very short telomeres were associated with AR/XLR or TINF2 compared with AD disease (OR 13.5, 95% CI 4.52-58.54, P < .01). Complex phenotypes (HH, RS, CS) showed significant lower telomere length z-scores than other DC/TBD patients combined (P < .01).
Ninety-six patients (41.6%) were children (<18 years) at diagnosis (Table 1). Childhood diagnoses occurred at a median 6.3 years of age (range 0 to 16.9) and were more likely associated with AR/XLR or TINF2 than AD disease (OR 9.2, 95% CI 4.5-20.11, P < .01, adjusted for sex). AD patients were the oldest with a median age of 36.1 years (range 0.7 to 69.4); AR/XLR were 11.3 years (range 0 to 45.9), and TINF2 patients were 8.1 years (range 0 to 71.6) at diagnosis.
Phenotypes and disease progression in the clinic cohort
The 91 clinic cohort participants were assessed at a median age of 23.8 years (range 1.4 to 69.4) by expert subspecialty clinicians (Table 2; Figure 2A-C). Patients with AR/XLR or TINF2 disease were more likely to have cytopenias requiring treatment, in addition to more neuropsychiatric, ophthalmologic, and dermatologic manifestations compared with AD disease. Median follow-up duration since the NIH visit was 7.1 years (range 0 to 17.1). During follow-up, 4 individuals (3 TINF2, 1 AR) progressed from 0-1 to 2-3 mucocutaneous triad features, and 9 advanced from nonsevere to severe BMF. Thirteen patients were diagnosed with PF on follow-up in addition to the 6 with PF at NIH visit (Figure 2A). Twelve (7 AD, 2 AR/XLR, 2 TINF2, 1 unknown) had not received HCT at time of PF diagnosis, while 7 developed PF following HCT (Figure 2B,C). Nine (1 AD, 6 AR/XLR, 2 TINF2) participants had PAVMs, including 8 following HCT and 4 cooccurring with PF. Three individuals had severe LD (fibrosis, cirrhosis, and/or portal hypertension) at NIH visit, and 8 patients subsequently developed LD. Seven (4 AR/XLR, 2 TINF2, 1 unknown) of these 11 had gastrointestinal vascular disease cooccurring with portal hypertension. Esophageal strictures were noted in 7 AR/XLR patients prior to or at NIH evaluation; 4 more (1 AD, 2 AR/XLR, 1 unknown) developed esophageal strictures during follow-up. Twenty patients reported nonspecific gastrointestinal problems including gastroesophageal reflux disease (GERD), gastritis, lactose intolerance, irritable bowel syndrome, delayed gastric emptying/gastroparesis, Barrett’s esophagitis, and ulcerative colitis.
Features of clinic cohort participants at the NIH Clinical Center evaluation
| Number of patients | Total, n (%) | AD, n (%) | AR/XLR, n (%) | TINF2, n (%) | Unknown, n (%) |
|---|---|---|---|---|---|
| 91 | 42 | 28 | 14 | 7 | |
| Age at NIH visit, median [range] | 23.8 [1.4-69.4] | 38.4 [1.5-69.4] | 14.5 [1.4-46.3] | 13.1 [2.1-31.4] | 13.1 [7.4-30.2] |
| Median follow-up in years since NIH visit [range] | 7.1 [0-17.1] | 7.1 [0-17.1] | 6.4 [0-14.5] | 12.6 [1.4-15.6] | 5.8 [0-8.5] |
| Consanguinity | 2 (2.2) | 0 | 2 (7.1) | 0 | 0 |
| Prematurity* | 20 (22) | 6 (14.3) | 8 (28.6) | 3 (21.4) | 3 (42.9) |
| Small for gestational age† [records available] | 26 [81] (32.1) | 4 [32] (12.5) | 16 [28] (57.1) | 3 [14] (21.4) | 3 [7] (42.9) |
| Short stature‡ | 6 (6.6) | 2 (4.8) | 3 (10.7) | 0 | 1 (14.3) |
| Number of triad features§ | |||||
| 0 | 38 (41.8) | 31 (73.8) | 5 (17.8) | 2 (14.2) | 0 |
| 1 | 14 (15.4) | 7 (16.7) | 3 (10.7) | 2 (14.2) | 2 (28.6) |
| 2 | 12 (13.2) | 1 (2.4) | 7 (25) | 1 (7.1) | 3 (42.9) |
| 3 | 27 (29.7) | 3 (7.1) | 13 (46.4) | 9 (64.3) | 2 (28.6) |
| BMFǁ | |||||
| None | 35 (38.5) | 26 (61.9) | 5 (17.9) | 1 (7.1) | 3 (42.9) |
| Nonsevere | 23 (25.3) | 9 (21.4) | 8 (28.6) | 4 (28.6) | 2 (28.6) |
| Severe | 33 (36.3) | 7 (16.7) | 15 (53.6) | 9 (64.3) | 2 (28.6) |
| Immunologic abnormality¶ [available records] | 30 [84] (35.7)# | 11 [40] (27.5) | 13 [27] (48.1) | 4 [11] (36.4) | 2 [6] (33.3) |
| Lymphocyte subset reduced** [available records] | 27 [77] (35.1) | 10 [39] (25.6) | 12 [23] (52.2) | 3 [9] (33.3) | 2 [6] (33.3) |
| Low IgM/IgG [available records] | 8 [82] (9.8) | 1 [38] (2.6) | 5 [27] (18.5) | 1 [11] (9.1) | 1 [6] (16.7) |
| Neurological | |||||
| Microcephaly [available records]†† | 17 [82] (20.7) | 2 [34] (5.9) | 10 [28] (35.7) | 3 [14] (21.4) | 2 [6] (33.3) |
| Cerebellar hypoplasia‡‡ [available imaging records] | 22 [59] (37.3) | 3 [17] (17.6) | 15 [25] (60) | 2 [10] (20) | 2 [7] (28.6) |
| Developmental delay | 26 (28.6) | 2 (4.8) | 20 (71.4) | 4 (28.6) | 0 |
| Ataxia | 14 (15.4) | 2 (4.8) | 9 (32.1) | 3 (21.4) | 0 |
| Ophthalmologic abnormalitiesa | 29 (31.9) | 4 (9.5) | 12 (42.9) | 10 (71.4) | 3 (42.9) |
| Epithelial: lacrimal duct stenosis, epiphora, blepharitis, trichiasis | 25 (27.5) | 3 (7.1) | 11 (39.3) | 9 (64.3) | 2 (28.6) |
| Retinal: retinopathy, retinal hemorrhage, retinal neovascularization/vascular sheathing [available records] | 7b [82] (8.5) | 1 [36] (2.8) | 2 [25] (8) | 3b [14] (21.4) | 1 [7] (14.3) |
| Taurodontism [total of dental consults] | 21 [70] (30) | 7 [31] (22.6) | 9 [23] (39.1) | 2 [11] (18.2) | 3 [5] (60) |
| Hearing loss [available records] | 4 [90] (4.4) | 1 [42] (2.4) | 1 [27] (3.7) | 1 [14] (7.1) | 1 [7] (14.3) |
| Congenital cardiac disease | 2 (2.2)c | 0 | 1 (3.6) | 0 | 1 (14.3) |
| Pulmonary fibrosis | 6 (6.6) | 5 (11.9) | 0 | 0 | 1 (14.3) |
| Prior to HCT | 6 (6.6) | 5 (11.9) | 0 | 0 | 1 (14.3) |
| Following HCT | 0 | 0 | 0 | 0 | 0 |
| Pulmonary arteriovenous malformations | 0 | 0 | 0 | 0 | 0 |
| Liver disease | 3 (3.3) | 0 | 2 (7.1) | 0 | 1 (14.3) |
| Portal hypertension | 2 (2.2) | 0 | 2 (7.1) | 0 | 0 |
| Liver fibrosis/cirrhosis | 1 (1.1) | 0 | 0 | 0 | 1 (14.3) |
| Gastrointestinal | |||||
| Esophageal strictures | 7 (7.7) | 0 | 7 (25) | 0 | 0 |
| Failure to thrive | 8 (8.8) | 0 | 6 (21.4) | 1 (7.1) | 1 (14.3) |
| Telangiectasiad | 1 (1.1) | 0 | 1 (3.6) | 0 | 0 |
| Genitourinary system | |||||
| Kidney structural abnormalitiese | 3 (3.3) | 1 (2.4) | 1 (3.6) | 1 (7.1) | 0 |
| Urethral stricture [number of males] | 6 [57] (10.5) | 0 [18] (0) | 3 [23] (13) | 2 [12] (16.7) | 1 [4] (25) |
| Undescended testis [number of males] | 4 [57] (7) | 2 [18] (11.1) | 2 [23] (8.7) | 0 [12] (0) | 0 [4] (0) |
| Endocrine system | |||||
| Hypothyroidism or diabetes mellitus, type 1 or 2 | 12 (13.2) | 6 (14.3) | 5 (17.9) | 0 (0) | 1 (14.3) |
| Skeletal system | |||||
| Avascular osteonecrosis | 5 (5.5) | 2 (4.8) | 2 (7.1) | 0 (0) | 1 (14.3) |
| Low bone density [number of DEXA scans] | 12 [38] (31.6) | 5 [14] (35.7) | 5 [13] (38.5) | 2 [7] (28.6) | 0 [4] (0) |
| Number of patients | Total, n (%) | AD, n (%) | AR/XLR, n (%) | TINF2, n (%) | Unknown, n (%) |
|---|---|---|---|---|---|
| 91 | 42 | 28 | 14 | 7 | |
| Age at NIH visit, median [range] | 23.8 [1.4-69.4] | 38.4 [1.5-69.4] | 14.5 [1.4-46.3] | 13.1 [2.1-31.4] | 13.1 [7.4-30.2] |
| Median follow-up in years since NIH visit [range] | 7.1 [0-17.1] | 7.1 [0-17.1] | 6.4 [0-14.5] | 12.6 [1.4-15.6] | 5.8 [0-8.5] |
| Consanguinity | 2 (2.2) | 0 | 2 (7.1) | 0 | 0 |
| Prematurity* | 20 (22) | 6 (14.3) | 8 (28.6) | 3 (21.4) | 3 (42.9) |
| Small for gestational age† [records available] | 26 [81] (32.1) | 4 [32] (12.5) | 16 [28] (57.1) | 3 [14] (21.4) | 3 [7] (42.9) |
| Short stature‡ | 6 (6.6) | 2 (4.8) | 3 (10.7) | 0 | 1 (14.3) |
| Number of triad features§ | |||||
| 0 | 38 (41.8) | 31 (73.8) | 5 (17.8) | 2 (14.2) | 0 |
| 1 | 14 (15.4) | 7 (16.7) | 3 (10.7) | 2 (14.2) | 2 (28.6) |
| 2 | 12 (13.2) | 1 (2.4) | 7 (25) | 1 (7.1) | 3 (42.9) |
| 3 | 27 (29.7) | 3 (7.1) | 13 (46.4) | 9 (64.3) | 2 (28.6) |
| BMFǁ | |||||
| None | 35 (38.5) | 26 (61.9) | 5 (17.9) | 1 (7.1) | 3 (42.9) |
| Nonsevere | 23 (25.3) | 9 (21.4) | 8 (28.6) | 4 (28.6) | 2 (28.6) |
| Severe | 33 (36.3) | 7 (16.7) | 15 (53.6) | 9 (64.3) | 2 (28.6) |
| Immunologic abnormality¶ [available records] | 30 [84] (35.7)# | 11 [40] (27.5) | 13 [27] (48.1) | 4 [11] (36.4) | 2 [6] (33.3) |
| Lymphocyte subset reduced** [available records] | 27 [77] (35.1) | 10 [39] (25.6) | 12 [23] (52.2) | 3 [9] (33.3) | 2 [6] (33.3) |
| Low IgM/IgG [available records] | 8 [82] (9.8) | 1 [38] (2.6) | 5 [27] (18.5) | 1 [11] (9.1) | 1 [6] (16.7) |
| Neurological | |||||
| Microcephaly [available records]†† | 17 [82] (20.7) | 2 [34] (5.9) | 10 [28] (35.7) | 3 [14] (21.4) | 2 [6] (33.3) |
| Cerebellar hypoplasia‡‡ [available imaging records] | 22 [59] (37.3) | 3 [17] (17.6) | 15 [25] (60) | 2 [10] (20) | 2 [7] (28.6) |
| Developmental delay | 26 (28.6) | 2 (4.8) | 20 (71.4) | 4 (28.6) | 0 |
| Ataxia | 14 (15.4) | 2 (4.8) | 9 (32.1) | 3 (21.4) | 0 |
| Ophthalmologic abnormalitiesa | 29 (31.9) | 4 (9.5) | 12 (42.9) | 10 (71.4) | 3 (42.9) |
| Epithelial: lacrimal duct stenosis, epiphora, blepharitis, trichiasis | 25 (27.5) | 3 (7.1) | 11 (39.3) | 9 (64.3) | 2 (28.6) |
| Retinal: retinopathy, retinal hemorrhage, retinal neovascularization/vascular sheathing [available records] | 7b [82] (8.5) | 1 [36] (2.8) | 2 [25] (8) | 3b [14] (21.4) | 1 [7] (14.3) |
| Taurodontism [total of dental consults] | 21 [70] (30) | 7 [31] (22.6) | 9 [23] (39.1) | 2 [11] (18.2) | 3 [5] (60) |
| Hearing loss [available records] | 4 [90] (4.4) | 1 [42] (2.4) | 1 [27] (3.7) | 1 [14] (7.1) | 1 [7] (14.3) |
| Congenital cardiac disease | 2 (2.2)c | 0 | 1 (3.6) | 0 | 1 (14.3) |
| Pulmonary fibrosis | 6 (6.6) | 5 (11.9) | 0 | 0 | 1 (14.3) |
| Prior to HCT | 6 (6.6) | 5 (11.9) | 0 | 0 | 1 (14.3) |
| Following HCT | 0 | 0 | 0 | 0 | 0 |
| Pulmonary arteriovenous malformations | 0 | 0 | 0 | 0 | 0 |
| Liver disease | 3 (3.3) | 0 | 2 (7.1) | 0 | 1 (14.3) |
| Portal hypertension | 2 (2.2) | 0 | 2 (7.1) | 0 | 0 |
| Liver fibrosis/cirrhosis | 1 (1.1) | 0 | 0 | 0 | 1 (14.3) |
| Gastrointestinal | |||||
| Esophageal strictures | 7 (7.7) | 0 | 7 (25) | 0 | 0 |
| Failure to thrive | 8 (8.8) | 0 | 6 (21.4) | 1 (7.1) | 1 (14.3) |
| Telangiectasiad | 1 (1.1) | 0 | 1 (3.6) | 0 | 0 |
| Genitourinary system | |||||
| Kidney structural abnormalitiese | 3 (3.3) | 1 (2.4) | 1 (3.6) | 1 (7.1) | 0 |
| Urethral stricture [number of males] | 6 [57] (10.5) | 0 [18] (0) | 3 [23] (13) | 2 [12] (16.7) | 1 [4] (25) |
| Undescended testis [number of males] | 4 [57] (7) | 2 [18] (11.1) | 2 [23] (8.7) | 0 [12] (0) | 0 [4] (0) |
| Endocrine system | |||||
| Hypothyroidism or diabetes mellitus, type 1 or 2 | 12 (13.2) | 6 (14.3) | 5 (17.9) | 0 (0) | 1 (14.3) |
| Skeletal system | |||||
| Avascular osteonecrosis | 5 (5.5) | 2 (4.8) | 2 (7.1) | 0 (0) | 1 (14.3) |
| Low bone density [number of DEXA scans] | 12 [38] (31.6) | 5 [14] (35.7) | 5 [13] (38.5) | 2 [7] (28.6) | 0 [4] (0) |
AD, autosomal dominant; AR, autosomal recessive; BMF, bone marrow failure; DEXA, dual energy X-ray absorptiometry; GI, gastrointestinal; HCT, hematopoietic cell transplantation; NIH, National Institutes of Health; XLR, X-linked recessive.
Prematurity: <37 gestational weeks.
Definition for small for gestational age: <37 gestational weeks: birthweight <tenth percentile (www.cdc.gov); ≥37 gestational weeks: <2500g.
Definition short stature: height <third percentile for age.
Triad features: mucocutaneous features associated with dyskeratosis congenita. Includes nail dysplasia, lacy/reticular skin pigmentation, leukoplakia.
n = 5 received HCT prior to study. Definition BMF severity: non-severe: absolute neutrophile count (ANC) 500-<1500/mm3, platelets 20 000-<150.000/mm3, and/or Hb ≥8g/dL to less than normal for age. Severe: transfusion dependency, and/or androgen treatment, and/or blood count: ANC <500/mm3, platelets <20 000/mm3, and/or Hb <8.0 g/dL. BMF was always considered severe if HCT had been performed, and/or MDS or leukemia had been diagnosed.
Immunologic diagnostics included lymphocyte subsets and/or immunoglobulin M (IgM) and G (IgG) levels. Immunologic abnormality refers to reduced levels in lymphocyte subsets and/or IgG and/or IgM levels according to the reference levels by the National Institutes of Health laboratory at time of study. Five patients with prior HCT and 1 with current chemotherapy (azacytidine) were excluded from the evaluation. Of the remaining, 84 participants had lymphocyte subsets and/or immunoglobulins done.
Of 30 patients with immunologic abnormalities, 15 had severe BMF and 1 received prednisone at time of study.
Reduced NK cell count n = 22, reduced CD19 B-cell count n = 14, reduced CD3 T-cell count n = 11, reduced CD4 T-cell count n = 12, reduced CD8 T-cell count n = 7.
Head circumference <5th percentile for age.
One patient was reported to have cerebellar hypoplasia on autopsy but was not diagnosed by brain imaging at time of study.
Nine patients were diagnosed with cataracts prior to time of study, which were age related in 5, congenital in 2 (siblings), coexistent to Coats retinopathy in 1, and in the context of Revesz syndrome in 1 case.
One patient had a history of HCT at time of ophthalmologic exam.
One patient with dilated cardiomyopathy and one patient with atrioventricular canal defect.
Esophageal varices diagnosed in the context of portal hypertension.
Structural abnormalities detected were: horseshoe kidney, bilateral renal cysts, duplication of renal collecting system and ureters.
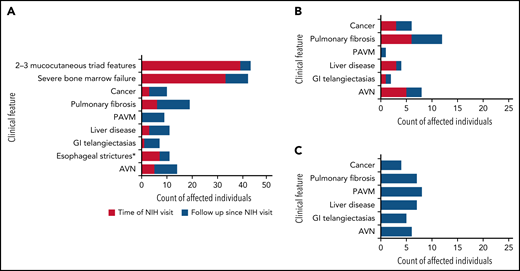
Clinical complications in the clinic cohort. (A) Development of clinical complications over time in 91 patients (clinic cohort). Red shaded areas show number of participants with clinical features (indicated on the y-axis) at time of evaluation at the National Institutes of Health. Blue shaded areas indicate the additional number of patients who developed these complications during follow-up. Mucocutaneous triad features include nail dysplasia, lacy reticular skin pigmentation, oral leukoplakia. Severe BMF: ANC <500/mm3, platelets <20 000/mm3, and/or Hb <8.0 g/dL OR cytopenia requiring treatment (HCT, regular platelet or red blood cell transfusions, or androgen treatment) and/or diagnosis of myelodysplastic syndrome or leukemia. (B) Clinical complications occurring with no prior hematopoietic cell transplantation. (C) Clinical complications occurring following HCT. AVN, avascular osteonecrosis of hip(s), knees, and/or shoulder(s); GI, gastrointestinal; PAVM, pulmonary arteriovenous malformation. *Esophageal strictures in 2 cases were diagnosed following HCT without history of gut GvHD.
Clinical complications in the clinic cohort. (A) Development of clinical complications over time in 91 patients (clinic cohort). Red shaded areas show number of participants with clinical features (indicated on the y-axis) at time of evaluation at the National Institutes of Health. Blue shaded areas indicate the additional number of patients who developed these complications during follow-up. Mucocutaneous triad features include nail dysplasia, lacy reticular skin pigmentation, oral leukoplakia. Severe BMF: ANC <500/mm3, platelets <20 000/mm3, and/or Hb <8.0 g/dL OR cytopenia requiring treatment (HCT, regular platelet or red blood cell transfusions, or androgen treatment) and/or diagnosis of myelodysplastic syndrome or leukemia. (B) Clinical complications occurring with no prior hematopoietic cell transplantation. (C) Clinical complications occurring following HCT. AVN, avascular osteonecrosis of hip(s), knees, and/or shoulder(s); GI, gastrointestinal; PAVM, pulmonary arteriovenous malformation. *Esophageal strictures in 2 cases were diagnosed following HCT without history of gut GvHD.
Five patients had a history of avascular osteonecrosis (AVN) of femoral and/or humoral head at the first visit, and 9 individuals developed AVN during follow-up (5 AD, 6 AR/XLR, 2 TINF2, 1 unknown). Six of these occurred following HCT (Figure 2C). One case of AVN of the mandible of a patient following radiotherapy for HNSCC was not included. Seven patients (3 AR/XLR, 3 TINF2, 1 unknown) reported fractures of long bones unrelated to AVNs following minor trauma. A tibial fracture occurred in 1 patient without prior HCT or intake of known predisposing medication. All other cases may have been related to prior androgen treatment and/or steroid intake during HCT.
Major disease manifestations in the combined clinic and field cohorts
As there were no statistically significant differences between the field and clinic cohort demographics, we combined data from both cohort participants (n = 231) to gain a better understanding of the major complications of DC/TBDs (Table 3). The median follow-up since diagnosis for the combined cohorts was 5.2 years (0 to 36.7). The median age of last contact or HCT was 42.9 years (range 2.2 to 82.2, n = 112) for AD, 19.8 years (range 0.9 to 54.2, n = 63) for AR/XLR, and 10.9 years (range 2.1 to 79.6, n = 25) for TINF2. Seventy patients received HCT with a median follow-up since HCT of 3.1 years (0 to 21.3) (Table 3).
Severe clinical complications in combined clinic and field cohort patients according to mode of inheritance
| Number of patients | Total, n (%) 231 | AD, n (%) 112 | AR/XLR, n (%) 63 | TINF2, n (%) 25 | Unknown, n (%) 31 |
|---|---|---|---|---|---|
| Number of patients with HCT | 70 | 20 | 27 | 15 | 8 |
| Median age at diagnosis in years [range]* | 19.4 [0-71.6] | 36.1 [0.7-69.4] | 11.3 [0-45.9] | 8.1 [0-71.6] | 13.2 [1.2-56.7] |
| Median age at follow-up in years [range] | 29.6 [1.3-82.2] | 42.9 [2.4- 82.2] | 21.1 [1.4-54.2] | 19.3 [4.6-79.6] | 18.2 [1.3-57.6] |
| Number deceased at last follow-up [median age at death, range] | 97 [29.6,1.3-82.2] | 31 [53.7, 2.4- 82.2] | 38 [22.7,1.4-54.2] | 14 [16.4, 4.6-79.6] | 14 [22.3, 1.3-57.6] |
| Severe BMF† | 111 (48.1) | 33 (29.5) | 44 (69.8) | 19 (76) | 15 (48.4) |
| Pulmonary disease | |||||
| Pulmonary fibrosis | |||||
| Prior to HCT | 33 (14.3) | 22 (19.6) | 6 (9.5) | 2 (8) | 3 (9.7) |
| Following HCT | 13 (18.6) | 1 (5) | 6 (22.2) | 5 (33.3) | 1 (12.5) |
| PAVM | |||||
| Prior to HCT | 1 (0.4) | 0 | 1 (1.6) | 0 | 0 |
| Following HCT | 11 (15.7) | 1 (5) | 6 (22.2) | 4 (26.7) | 0 |
| Severe liver disease‡ | |||||
| Prior to HCT | 12 (5.2) | 2 (1.8) | 6 (9.5) | 0 | 4 (12.9) |
| Following HCT | 9 (12.9) | 2 (10) | 3 (11.1) | 3 (20) | 1 (12.5) |
| Gastrointestinal complications | |||||
| Esophageal strictures | 23§ (10) | 2 (1.8) | 14 (22.2) | 0 | 7 (22.6) |
| GI telangiectasia | |||||
| Prior to HCT | 9 (3.9) | 2 (1.8) | 5 (7.9) | 0 | 2 (6.5) |
| Following HCT | 6 (8.6) | 1 (5) | 2 (7.4) | 2 (13.3) | 1 (12.5) |
| Avascular osteonecrosis | |||||
| Prior to HCT | 20 (8.7)ǁ | 10 (8.9) | 7 (11.1) | 2 (8) | 1 (3.2) |
| Following HCT | 9 (12.9) | 3 (15) | 4 (14.8) | 2 (13.3) | 0 |
| Myelodysplastic syndrome | 17 (7.4) | 12 (10.7) | 4 (6.3) | 0 | 1 (3.2) |
| Cancer¶ | |||||
| Prior to HCT | 27 (11.7) | 13 (11.6) | 8 (12.7) | 1 (4) | 5 (16.1) |
| Leukemia | 7 (3) | 5 (4.5) | 1 (1.6) | 0 | 1 (3.2) |
| Solid tumor# | 20 (8.7) | 8 (7.1) | 7 (11.1) | 1 (4) | 4 (12.9) |
| Following HCT | 5 (7.1) | 0 | 2 (7.4) | 3 (20) | 0 |
| Number of patients | Total, n (%) 231 | AD, n (%) 112 | AR/XLR, n (%) 63 | TINF2, n (%) 25 | Unknown, n (%) 31 |
|---|---|---|---|---|---|
| Number of patients with HCT | 70 | 20 | 27 | 15 | 8 |
| Median age at diagnosis in years [range]* | 19.4 [0-71.6] | 36.1 [0.7-69.4] | 11.3 [0-45.9] | 8.1 [0-71.6] | 13.2 [1.2-56.7] |
| Median age at follow-up in years [range] | 29.6 [1.3-82.2] | 42.9 [2.4- 82.2] | 21.1 [1.4-54.2] | 19.3 [4.6-79.6] | 18.2 [1.3-57.6] |
| Number deceased at last follow-up [median age at death, range] | 97 [29.6,1.3-82.2] | 31 [53.7, 2.4- 82.2] | 38 [22.7,1.4-54.2] | 14 [16.4, 4.6-79.6] | 14 [22.3, 1.3-57.6] |
| Severe BMF† | 111 (48.1) | 33 (29.5) | 44 (69.8) | 19 (76) | 15 (48.4) |
| Pulmonary disease | |||||
| Pulmonary fibrosis | |||||
| Prior to HCT | 33 (14.3) | 22 (19.6) | 6 (9.5) | 2 (8) | 3 (9.7) |
| Following HCT | 13 (18.6) | 1 (5) | 6 (22.2) | 5 (33.3) | 1 (12.5) |
| PAVM | |||||
| Prior to HCT | 1 (0.4) | 0 | 1 (1.6) | 0 | 0 |
| Following HCT | 11 (15.7) | 1 (5) | 6 (22.2) | 4 (26.7) | 0 |
| Severe liver disease‡ | |||||
| Prior to HCT | 12 (5.2) | 2 (1.8) | 6 (9.5) | 0 | 4 (12.9) |
| Following HCT | 9 (12.9) | 2 (10) | 3 (11.1) | 3 (20) | 1 (12.5) |
| Gastrointestinal complications | |||||
| Esophageal strictures | 23§ (10) | 2 (1.8) | 14 (22.2) | 0 | 7 (22.6) |
| GI telangiectasia | |||||
| Prior to HCT | 9 (3.9) | 2 (1.8) | 5 (7.9) | 0 | 2 (6.5) |
| Following HCT | 6 (8.6) | 1 (5) | 2 (7.4) | 2 (13.3) | 1 (12.5) |
| Avascular osteonecrosis | |||||
| Prior to HCT | 20 (8.7)ǁ | 10 (8.9) | 7 (11.1) | 2 (8) | 1 (3.2) |
| Following HCT | 9 (12.9) | 3 (15) | 4 (14.8) | 2 (13.3) | 0 |
| Myelodysplastic syndrome | 17 (7.4) | 12 (10.7) | 4 (6.3) | 0 | 1 (3.2) |
| Cancer¶ | |||||
| Prior to HCT | 27 (11.7) | 13 (11.6) | 8 (12.7) | 1 (4) | 5 (16.1) |
| Leukemia | 7 (3) | 5 (4.5) | 1 (1.6) | 0 | 1 (3.2) |
| Solid tumor# | 20 (8.7) | 8 (7.1) | 7 (11.1) | 1 (4) | 4 (12.9) |
| Following HCT | 5 (7.1) | 0 | 2 (7.4) | 3 (20) | 0 |
See Table 2 for abbreviation definitions.
The diagnosis was established postmortem for 24 patients: they were not included in the calculation of age at diagnosis.
Severe: transfusion dependency, and/or androgen treatment, and/or blood count: ANC <500/mm3, platelets <20 000/mm3, and/or Hb <8.0 g/dL. BMF was always considered severe if HCT had been performed, and/or MDS or leukemia had been diagnosed.
Definition of severe liver disease: portal hypertension and/or liver cirrhosis/fibrosis.
One reported in the context of severe gut GvHD. One adult patient reported to have pyloric stenosis several years after HCT. Three patients had esophageal strictures mentioned in medical reports following HCT without specifying a GvHD diagnosis.
Two reported previous steroid treatment over several years (unknown dose). One patient received immunosuppression for lung transplant before developing hip avascular osteonecrosis. Not listed: 1 patient had osteonecrosis of the mandible following radiotherapy for head and neck squamous cell carcinoma.
Excluding 13 patients diagnosed with nonmelanoma skin cancer (squamous cell carcinoma or basal cell carcinoma), 1 following HCT.
Cancer cases included 11 head and neck cancers, 3 esophageal cancers, 3 anal/rectal cancers, 3 non-Hodgin lymphomas, 1 endometroid cancer, 1 cervical cancer. Two patients reported the occurrence of more than 1 solid tumor entity.
Severe BMF developed in 111 (48.1%) patients, and 70 (30.3%) underwent HCT at a median age of 15.4 years (range 0.9 to 63.1). The risk of developing severe BMF in AR/XLR or TINF2 was significantly higher compared with AD (OR 5.5, 95% CI 2.87-11.08, P < .01; OR 7.6, 95% CI 2.92-22.4, P < .01, respectively). Of note, in AD TERC patients a significantly higher prevalence of severe BMF (56.7%) was reported than in AD RTEL1 (13.8%, P < .01) or AD TERT (25.5%, P < .01) (supplemental Table 7).
PF developed in 33 adults without prior HCT occurring in 19.6% of those with AD disease compared with 9.5% in AR/XLR (P = .09) and 8% in TINF2 (P = .25) (Table 3). Following HCT, PF was more common in AR/XLR (22.2%) and TINF2 (33.3%) than in AD disease (5%, P = .21 and P = .07, respectively) (Table 3). Eight of 13 post-HCT PF cases manifested in childhood. Twelve patients developed PAVMs (n = 5 with coexistent PF), including the 7 previously published.13 Eleven cases of PAVMs occurred after HCT, with 10 in AR/XLR or TINF2 patients (Table 3).
LD (portal hypertension and/or liver cirrhosis/fibrosis) was reported in 12 patients without prior HCT (Table 3) and was significantly more frequent in AR/XLR compared with AD (OR 11.6, 95% CI 1.84-138.45, P = .02, adjusted for pediatric vs adult at DC/TBD diagnosis and sex). Nine patients (2 AD, 3 AR/XLR, 3 TINF2, 1 unknown) developed severe LD following HCT without a history of liver graft-versus-host disease (GvHD). Hepatopulmonary syndrome (HPS) without prior HCT was reported in 2 patients (1 AD, 1 AR/XLR). Four had HPS (2 AR/XLR, 2 TINF2) following HCT (median time since HCT 5.7 years, range 4.4 to 7.4) with 3 diagnosed <18 years of age.
Gastrointestinal telangiectasias cooccurring with portal hypertension were diagnosed in 9 patients without prior HCT, predominantly in AR/XLR patients (Table 3). Six patients were diagnosed with GI telangiectasias after HCT. Esophageal strictures occurred in 10% and were more common in AR/XLR disease compared with TINF2 and AD disease combined (OR 2.7, 95% CI 1.05-6.87, P = .04, adjusted for pediatric vs adult at DC/TBD diagnosis). Five patients reported esophageal stricture after HCT; 1 also had severe gut GvHD.
AVN of femoral and/or humoral head was frequent, occurring in 29 patients, with 28 having at least 1 femoral head affected (Table 3). Twenty individuals (8.7%) developed AVN without prior HCT (median age 27.1 years, range 6.3 to 56.1). The prevalence of AVN was similar in AD (8.9%), AR/XLR (11.1%), and TINF2 (8%) in individuals with no prior HCT or following HCT (15%, 14.8%, and 13.3%, respectively) (Table 3).
Malignancies
Twenty-seven patients (11.7%) with no prior HCT developed a first cancer at a median age of 40.5 years (range 18.2 to 67.5) (Table 3). Seven patients had leukemia. The most frequent nonhematologic malignancy was HNSCC (n = 11). Compared with AD disease, AR/XLR had a hazard ratio for any cancer of 8 (95% CI 1.93-33.51, adjusted for sex and pediatric vs adult at DC/TBD diagnosis). One patient with TINF2 had non-Hodgkin lymphoma (NHL) without preceding HCT. MDS was reported in 10.7% of AD patients, predominantly TERC, 6.3% of AR/XLR, and none of the TINF2 patients (n = 1 with unknown genotype). The median age at MDS diagnosis was 51.4 years (range 5.9 to 66); 53.2 years (range 26.6 to 66) in AD compared with 16.8 years (range 5.9 to 47.5) in AR/XLR patients (P = .01). Five patients (3 TINF2, 2 AR/XLR) developed solid malignancies after HCT. The median age at cancer diagnosis was 18.8 years (range 10.5 to 32.7), and median time since HCT was 4.6 years (range 1.8 to 14.6). Additionally, 3 patients developed posttransplant lymphoproliferative disease at a median of 4.6 years (0.2 to 5.5) from HCT.
Long-term outcomes in the combined clinic and field cohorts
Mortality
At last follow-up, 97 patients (42%) were deceased due to lung disease (PF/PAVM, 27.8%), cancer (15.5%), or BMF (15.5%). Seventeen patients who underwent HCT for BMF (n = 10), MDS (n = 3), AML (n = 3), or immunodeficiency (n = 1) died due to HCT-related complications. One patient died during AML-induction treatment, and 1 succumbed to complications following lung and liver transplant.
The most common reported cause of death in the 31 deceased AD patients was pulmonary disease (PF and/or PAVM, 38.7%). BMF (23.7%) and pulmonary disease (PF/PAVM, 23.7%) equally contributed to death in AR/XLR patients (n = 38). Three of 14 deaths in TINF2 patients were due to malignancies following HCT (supplemental Table 8).
The median OS age for the combined cohorts was 52.8 years (95% CI 45.5-57.6), with significantly lower survival in males vs females (P < .01) (Figure 3A), even after exclusion of patients with XLR disease. The median OS in patients with AD disease was 64.9 years (95% CI 59.8-67.6). It was 31.8 years (95% CI 24.2-37.4) in AR/XLR, 37.9 years (95% CI 13.5-47.3) in TINF2, and 27.8 years (95% CI 19.1-39.1) in those with unknown genotype (Figure 3B). AD disease had a significantly better OS than AR/XLR and TINF2 disease combined (P < .01), while patients with AR/XLR and TINF2 disease had similar survival probabilities (P = .33) (Figure 3B,C). The association remained similar after accounting for TL <first percentile (AR/XLR/TINF2 vs AD: HR 4.8, P < .01) (supplemental Table 9). Of note, when comparing 29 AD-RTEL1 to 15 AR-RTEL1, Kaplan Meier estimates of OS were significantly different as well (P < .01) (supplemental Figure 2).
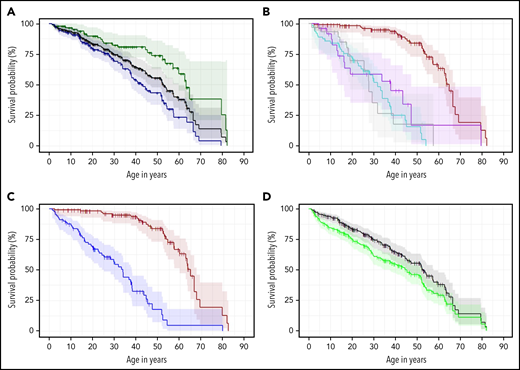
Kaplan Meier estimates of the combined field and clinic cohorts. (A) OS of combined cohorts: OS of complete cohort (field and clinic cohort, n = 231, black line) and divided by sex: 87 females (green line) and 144 males (blue line), P < .01. (B) OS according to inheritance pattern. Red line shows autosomal dominant/nonTINF2 (AD, n = 112); turquoise line shows autosomal recessive/X-linked recessive (AR/XLR, n = 63). Purple line indicates disease due to TINF2 (n = 25), and gray shows 31 patients with unknown genotype (AD vs AR/XLR P < .01, AD vs TINF2 P < .01, AR/XLR vs TINF2 P = .33). (C) OS of AD-nonTINF2 vs all other inheritance pattern groups: nonTINF2 AD disease (dark red, n = 112) vs all other inheritance pattern groups (AR/XLR/TINF2, blue, n = 88 P < .01). (D) OS compared with transplant-free survival. OS of 231 patients with telomere biology disorders (black line), with 77 of 231 receiving either HCT, liver transplant, and/or lung transplant. Transplant-free survival is indicated by the green line. One patient receiving a kidney transplant was not included in the calculations. Observed survival (stair-step lines) are shown; shaded areas show 95% confidence intervals. AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive.
Kaplan Meier estimates of the combined field and clinic cohorts. (A) OS of combined cohorts: OS of complete cohort (field and clinic cohort, n = 231, black line) and divided by sex: 87 females (green line) and 144 males (blue line), P < .01. (B) OS according to inheritance pattern. Red line shows autosomal dominant/nonTINF2 (AD, n = 112); turquoise line shows autosomal recessive/X-linked recessive (AR/XLR, n = 63). Purple line indicates disease due to TINF2 (n = 25), and gray shows 31 patients with unknown genotype (AD vs AR/XLR P < .01, AD vs TINF2 P < .01, AR/XLR vs TINF2 P = .33). (C) OS of AD-nonTINF2 vs all other inheritance pattern groups: nonTINF2 AD disease (dark red, n = 112) vs all other inheritance pattern groups (AR/XLR/TINF2, blue, n = 88 P < .01). (D) OS compared with transplant-free survival. OS of 231 patients with telomere biology disorders (black line), with 77 of 231 receiving either HCT, liver transplant, and/or lung transplant. Transplant-free survival is indicated by the green line. One patient receiving a kidney transplant was not included in the calculations. Observed survival (stair-step lines) are shown; shaded areas show 95% confidence intervals. AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive.
We conducted additional survival analysis excluding the 25 heterozygous family members of AR index patients from the AD subgroup: the OS in the AD group (n = 87) remained significantly better than in AR/XLR and TINF2 disease (P < .01) (supplemental Figure 3).
HCT was performed in 17.9% of all AD patients (median age 26.1 years, range 2.2 to 63.1) compared with 60% of TINF2 (median age 5.7 years, range 2.1 to 47.3) and 42.9% (16.8 years, range 0.9 to 34.7) of AR/XLR patients (AD vs TINF2: P < .01; AD vs AR/XLR: P < .01; TINF2 vs AR/XLR: P = .16) (Table 3).
Eight individuals (5 AD, 2 TINF2, 1 unknown genotype) had pulmonary disease (PF and/or HPS), leading to lung transplantation (median age 51.4 years, range 13.1 to 62.4). Two had a prior HCT; the time between HCT and lung transplantation was 4.5 and 8.9 years. Four (3 AR/XLR, 1 AD) of the 21 patients with severe LD received liver transplants (median age 27.8 years, range 21 to 56.7). Two had prior HCT and received liver transplants 6.5 and 11.9 years after HCT. Five patients received multiple organ transplants (n = 2 HCT then lung transplant, n = 2 HCT then liver transplant, n = 1 combined lung and liver transplant). The overall median HCT, liver, or lung TFS was 45.3 years (95% CI 37.4-52.1) (Figure 3D). The survival was significantly lower for the combined AR/XLR and TINF2 patients than for AD patients (P < .01, median age 22.7 [range 17.1 to 29.4] vs 62.7 [range 53.7 to 66.6] years).
The 5-year post HCT survival was 58% for the 70 patients who underwent HCT. The median follow-up for the 8 patients who underwent lung transplant was 3.1 years (0.4 to 8.6); 3 were alive 0.4 (n = 2) and 8.6 (n = 1) years after transplant. For liver transplant patients (n = 4), follow-up time since liver transplant was very short (<6 months) with 3 alive at last contact.
Discussion
Systematic, detailed phenotyping of 231 individuals with DC/TBDs identified clinically relevant differences in phenotypes and outcomes based on genotype. The longitudinal prospective cohort study design made it possible to describe the progression of DC/TBDs over several years in individuals of all ages. Our genotype-phenotype analysis builds a foundation for stratifying the care of patients with DC/TBDs by mode of inheritance.
The OS of patients with DC/TBDs was directly related to the mode of inheritance. We found significantly better survival in AD compared with AR/XLR or TINF2-associated DC/TBDs, likely due to the fewer medical problems associated with AD disease and/or the shortest telomeres occurring in the AR/XLR/TINF2 patients.27 AR/XLR and TINF2 showed similar survival probabilities, with the caveat that the TINF2 subgroup was distinctly smaller (n = 25) than the AR/XLR group (n = 63). Overall, AR/XLR and TINF2 patients were more likely to have multiorgan disease at younger ages than AD patients. This is consistent with previously published observations that patients with DKC1 and TINF2 pathogenic variants are usually younger and have more features than those with heterozygous TERC or TERT variants.5,6,16,42 We specifically report a higher prevalence of the mucocutaneous triad, neurologic abnormalities, severe BMF, LD, HPS, PAVM, and gastrointestinal bleeding in patients with AR/XLR or TINF2 DC/TBDs.
We previously reported a high prevalence of low bone mineral density in patients with inherited BMF syndromes, including DC.43 However, that study and others are limited because conventional DEXA scans are not reliable in predicting pediatric fracture risk.44 Osteoporosis, glucocorticoid intake, iron overload, and/or HCT contribute to fracture risk and may explain our observations of fractures unrelated to AVN in DC/TBDs. The fractures could also be due to primary disease; for example, CS patients have fracture tendency due to osteopenia.45-48
Similar to prior reports, PAVM and gastrointestinal telangiectasias were frequent findings in our cohort and often followed HCT.13,14,29 Notably, gastrointestinal vascular changes occurred only in patients with portal hypertension, whereas PAVM also occurred in patients without HPS or other evidence of LD.13 It is possible that PAVMs develop independent of LD as part of a yet-to-be-discovered connection between short telomeres and the vascular endothelium.29
Monoallelic TERT, TERC, RTEL1, and PARN pathogenic variants have been connected with isolated pulmonary disease, including PF and emphysema.34,48-53 Similarly, we found PF with no prior HCT primarily in AD patients, however, this finding was not significant. In our cohort, AR/XLR/TINF2 patients were younger at last contact than AD patients, and due to high mortality early in life, they may not reach the age to develop typical adult-onset complications. Following HCT, PF was reported more frequently in AR/XLR and TINF2, consistent with our previous study focusing on pulmonary disease in DC/TBD,11 although statistical significance was not reached, possibly due to small sample size.
HPS was previously reported as a main cause of pulmonary disease in younger TBD patients.54 Our study did not confirm this finding in younger AR/XLR/TINF2 patients, which may be due to different study ascertainment methods and/or statistical power.54 Isolated LD in TBDs has primarily been reported in the context of heterozygous TERT, RTEL1, or TERC pathogenic variants.9,55-57 However, in our cohort, LD was more prevalent in AR/XLR and TINF2-associated DC/TBD, possibly because we focused on severe LD (portal hypertension, liver cirrhosis/fibrosis, +/−HPS) and not milder liver involvement such as elevated transaminases or ultrasound abnormalities. Severe LD following HCT in patients with DC/TBDs has recently been identified as a major complication and might require, as was the case in 2 of our patients, liver transplant.58-60
Multiorgan transplants in DC/TBD individuals have been increasingly reported.61,62 In our cohort, 4 individuals had either lung or liver transplant following HCT, and 1 had a combined lung/liver transplant. The necessity of sequential lung or liver transplants following HCT highlights both the multiorgan involvement and risk of therapy-related toxicity in TBDs.63,64
Previous studies aiming to describe DC/TBD phenotypes have relied on smaller sample sizes, literature review, less genetically diverse cohorts, or limited registry data.42,65,66 Our combined field and clinic cohort approach, along with longitudinal follow-up, allowed for more detailed characterization of patients with DC/TBDs. An important strength of this study is the single-center, detailed phenotype evaluations of the clinic cohort participants. Like all observational studies, there are some limitations. Participants enroll in the IBMFS study at the NCI, and thus ascertainment may be biased toward those with BMF, cancer, and/or more complex phenotypes. Data collection for the field cohort was limited to patient reports and retrospective review of medical records, which may have led to underreporting of clinical findings, specifically of complications such as HPS, PAVMs, and gastrointestinal telangiectasias.
Our study provides a rationale for a genotype-based approach to DC/TBDs instead of a potentially highly variable phenotype-based approach. We recognize that grouping patients by inheritance pattern limits analyses by specific gene, but the limitations of small sample size preclude more detailed analyses. The AD subgroup was biased toward AD RTEL1/TERT/TERC individuals with only a few monoallelic ACD and PARN cases. A potential subgroup to be considered separately in future studies is BMF and MDS in patients with AD TERC. Biallelic RTEL1 and X-linked DKC1 variants represented the largest component of the AR/XLR subgroup. Consistent with prior studies, we found very short telomeres (<first percentile for age) to be more common in AR/XLR or TINF2 disease compared with AD.22,27,49 This association between the mode of inheritance and both phenotype and TL suggests that the clinical impact of genotypes could be mediated by the degree to which telomeres are shortened.27 TL did explain part of the associations between the clinical complications and inheritance patterns in our study, but the associations between the inheritance patterns and phenotypes remained statistically significant when adjusting for TL. To capture the total effect of genotype on phenotype, we subsequently focused on genotype-phenotype associations without adjusting for TL.
Conclusion
This large, longitudinal cohort study of DC/TBDs systematically evaluated the mode of inheritance and its relationship to the clinical course of these complex disorders. The presence of clinically significant differences in survival by gene and mode of inheritance illustrates that it may be possible to tailor clinical management and disease surveillance decisions in DC/TBDs according to the inheritance pattern. Patients with AR/XLR and TINF2-associated TBD need to be followed for early-onset BMF and multisystem disease, whereas AD patients should be educated about the possibility of late-onset disease. Future large studies to compare the clinical outcomes by gene, variant, and TL are warranted to advance understanding of the biology and clinical impact of variants in DC/TBD-associated genes.
Acknowledgments
The authors thank the study participants and their families for their valuable contributions. The authors acknowledge the NIH Clinical Center consultants from audiology, cardiology, dentistry, dermatology, gastroenterology/hepatology, imaging, neurology, ophthalmology, pulmonology, and psychiatry for subspeciality clinical evaluations and investigations of the participants, and also thank the participants’ referring clinicians.
This work was supported by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, grant number ZIA CP010190-20, and fellowship support for M.R.N. from the Mildred-Scheel-Postdoctoral Fellowship Program by the German Cancer Aid (70113506). Outstanding study support was provided by Ann Carr, Lisa Leathwood, and Maureen Risch of Westat, Inc. under contract HHN261201700004C with the NCI.
Authorship
Contribution: M.R.N., S.B., and N.G. collected patient data; M.R.N. and R.A. performed data analysis; M.R.N., N.G., B.P.A., and S.A.S. were involved in planning the project; M.R.N. and L.J.M. performed genetic data analysis; M.R.N. generated figures and tables and wrote the manuscript; M.R.N., N.G., and S.A.S. led the project; and all authors participated in discussions and interpretation of the data and results.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sharon A. Savage, Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, 9609 Medical Center Dr, MSC 9772, Rockville, MD 20892-9772; e-mail: savagesh@mail.nih.gov.
The deidentified data generated in this study are available upon reasonable request to Sharon A. Savage, provided the request is consistent with the study consent documents.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal