

In this issue of Blood, Cebo et al1 demonstrate that the platelet chemokine (C-X-C motif) receptor 7 (CXCR7) plays a novel role in regulating the platelet lipidome, and through the generation of “antithrombotic” lipid species, CXCR7 ligation can regulate thromboinflammatory platelet functional responses.
The quest for novel therapeutic strategies for the treatment of cardiovascular disease (CVD) represents an ongoing unmet clinical need because of the significant global burden of CVD. One of the fundamental mechanisms underlying the shortcomings of current therapies for CVD is the fact that thromboinflammation plays a key role in driving the thrombotic response; however, this is not abrogated by current antithrombotic therapies. Thromboinflammation reflects the bidirectional crosstalk between thrombosis and inflammation and is implicated in a broad range of clinical conditions, including CVD, sepsis, ischemia reperfusion injury, and coronavirus disease 2019 (COVID-19).2,3 Platelets are well recognized as central players mediating thromboinflammation owing to their expression of a large repertoire of receptors essential for adhesion and immune function, in conjunction with their ability to produce and release a range of cytokines and chemokines resulting in leukocyte recruitment and activation.2
In addition to releasing a number of important inflammatory mediators, such as chemokine (C-X-C motif) ligand 4 (CXCL4)/platelet factor 4, chemokine (C-C motif) receptor 5, CXCL12, migration inhibitory factor (MIF), interleukin-1β (IL-1β), and transforming growth factor-β, platelets also express the chemokine receptors, CXCR7 and CXCR4.4,5 As such, significant attention has recently been focused on how these chemokine receptors modulate the thrombotic and inflammatory function of platelets. Previous work has demonstrated that the interaction between CXCR7 with its 2 ligands, CXCL12 and MIF, promotes platelet lifespan and mediates an antithrombotic effect by inhibiting phosphatidylserine (PS) exposure.4 Moreover, platelet CXCR7 expression levels increase after myocardial infarction and correlate with an improved prognosis.6 However, the precise mechanisms governing how increased platelet CXCR7 expression may confer benefit in the setting of myocardial infarction and how this may be exploited therapeutically have remained elusive.
In this context, the study by Cebo and colleagues now links these critical observations and defines how platelet CXCR7 can regulate the thromboinflammatory function of platelets (see figure). Utilizing platelets from a large cohort of patients with CVD, the authors highlight the protective effect of increased platelet CXCR7 expression by demonstrating that increased platelet CXCR7 expression correlates with a reduction in platelet aggregation in response to a range of platelet agonists. In order to demonstrate that platelet CXCR7 plays a direct role in modulating platelet function, the authors use a CXCR7-specific agonist (VUF11207) to show that CXCR7 signaling significantly attenuates platelet thrombus formation under shear and inhibits soluble agonist activation ex vivo. These findings were recapitulated utilizing in vivo mouse models of myocardial infarction and arterial thrombosis and highlight that, similar to humans, mouse platelets increase CXCR7 expression after myocardial infarction. Furthermore, CXCR7 agonism results in a reduction in infarct size, reduced platelet activation, and reduced platelet-leukocyte aggregate formation in vivo. In accordance with the hypothesis that CXCR7 modulates the thromboinflammatory response, CXCR7 agonist treatment significantly alters the expression of proinflammatory mediators after myocardial infarction and inhibits the release of proinflammatory cytokines from thrombin-activated platelets. Indeed, treatment of mice with a CXCR7 agonist was found to be associated with a marked reduction in the levels of plasma cytokines, including IL-1α, interferon-γ, tumor necrosis factor-α, monocyte chemoattractant protein 1, IL-1β, and IL-6 after myocardial infarction. Moreover, although the treatment of mice with the CXCR7 agonist afforded protection from occlusive thrombus formation, no bleeding phenotype was observed by measuring tail bleeding time or basic coagulation parameters. This finding will require further investigation, as the tail bleeding time described in the current study does not correlate with the bleeding risk of antithrombotics.7 Indeed, it is noteworthy that CXCR7 agonist treatment inhibits platelet PS exposure, which is significant because the rare platelet function defect (Scott syndrome) associated with defective platelet PS externalization is associated with reduced thrombin generation and increased bleeding.8
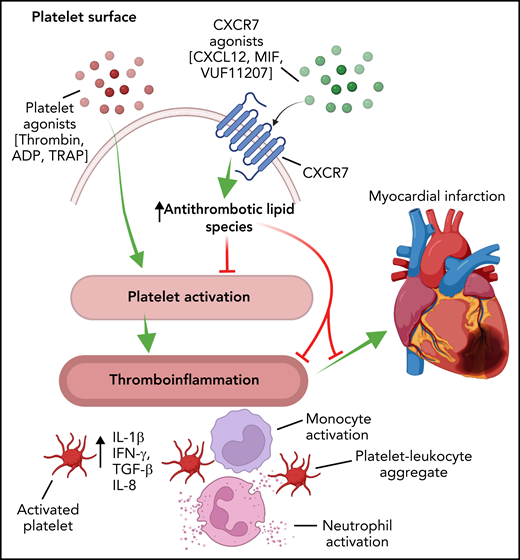
Schematic representation demonstrating that the activation of platelets elicits a thromboinflammatory response that is manifested by the release of cytokines, leukocyte activation, and the formation of platelet-leukocyte aggregates. These factors contribute to the pathogenesis of CVD, including myocardial infarction. The activation of platelet CXCR7 alters the platelet lipidome and generates lipid species that inhibit the thromboinflammatory function of platelets, thus affording protection from thrombosis and myocardial infarction. IFN-γ, interferon-γ; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α. Figure created with BioRender.com.
Schematic representation demonstrating that the activation of platelets elicits a thromboinflammatory response that is manifested by the release of cytokines, leukocyte activation, and the formation of platelet-leukocyte aggregates. These factors contribute to the pathogenesis of CVD, including myocardial infarction. The activation of platelet CXCR7 alters the platelet lipidome and generates lipid species that inhibit the thromboinflammatory function of platelets, thus affording protection from thrombosis and myocardial infarction. IFN-γ, interferon-γ; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α. Figure created with BioRender.com.
To afford mechanistic insights regarding the protective effects of CXCR7 agonist treatment in the context of thromboinflammation, the authors employed untargeted and targeted lipidomic analysis of CXCR7-treated platelets. Here they elegantly demonstrate that ligation of platelet CXCR7 alters the platelet lipidome, independent of antiplatelet drugs and statins, such that the platelet lipidome displays a reduction in a number of important lipid species known to be associated with prothrombotic and atherogenic roles, including lysophosphatidylinositol, lysophosphatidylcholine, diacylglycerol, thromboxane A2, and 12-lipoxygenase-LOX-(12-HETE).9 Concurrently, CXCR7 treatment of platelets was associated with an increase of platelet-derived 12-hydroxyeicosatrienoic acid, which, consistent with previous work, inhibits intracellular calcium mobilization and thus platelet activation via activation of the prostacyclin (IP) receptor.10
In summary, this study highlights the platelet CXCR7 signaling axis as a potential novel strategy to target thromboinflammation. Although the current work principally focuses on CVD, the ability to therapeutically target thromboinflammation is likely to have broad clinical relevance across a number of important clinical entities. However, it must be noted that several important outstanding questions remain. First, the role of platelet CXCR7 in other thromboinflammatory diseases, such as COVID-19, sepsis, and ischemia reperfusion injury, warrants further investigation. Moreover, CXCR7 is widely expressed on many tissues, including the myocardium and endothelium, and therefore, it remains to be established whether the impressive in vivo protection afforded in these studies relate to platelet-specific CXCR7 signaling or to the systemic effects of CXCR7 activation. These data will be critical in ultimately elucidating the true therapeutic potential of targeting platelet CXCR7 in the ongoing quest for novel therapeutic approaches for the treatment of thromboinflammation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal