

Key Points
CRISPR-AAV achieves efficient targeted insertion of MAGT1 in XMEN patient hematopoietic stem cells and lymphocytes.
MAGT1 correction restores NKG2D expression on gene-edited XMEN CD8+ T and NK cells.

Abstract
XMEN disease, defined as “X-linked MAGT1 deficiency with increased susceptibility to Epstein-Barr virus infection and N-linked glycosylation defect,” is a recently described primary immunodeficiency marked by defective T cells and natural killer (NK) cells. Unfortunately, a potentially curative hematopoietic stem cell transplantation is associated with high mortality rates. We sought to develop an ex vivo targeted gene therapy approach for patients with XMEN using a CRISPR/Cas9 adeno-associated vector (AAV) to insert a therapeutic MAGT1 gene at the constitutive locus under the regulation of the endogenous promoter. Clinical translation of CRISPR/Cas9 AAV-targeted gene editing (GE) is hampered by low engraftable gene-edited hematopoietic stem and progenitor cells (HSPCs). Here, we optimized GE conditions by transient enhancement of homology-directed repair while suppressing AAV-associated DNA damage response to achieve highly efficient (>60%) genetic correction in engrafting XMEN HSPCs in transplanted mice. Restored MAGT1 glycosylation function in human NK and CD8+ T cells restored NK group 2 member D (NKG2D) expression and function in XMEN lymphocytes for potential treatment of infections, and it corrected HSPCs for long-term gene therapy, thus offering 2 efficient therapeutic options for XMEN poised for clinical translation.
Introduction
Defective magnesium transporter 1 (MAGT1) impairs N-linked glycosylation of some key immune proteins1-3 such as the natural killer (NK) group 2 member D (NKG2D) receptor expression on CD8+ T cells and NK cells that causes a primary immunodeficiency (PID) XMEN disease. XMEN disease is defined as “X-linked MAGT1 deficiency with increased susceptibility to Epstein-Barr virus (EBV) infection and N-linked glycosylation defect”3-7 and is associated with lymphomas.8-11 Transfection of XMEN lymphocytes with exogenous MAGT1 messenger RNA (mRNA) reversed defective glycosylation in lymphocytes and restored NKG2D expression,3,12 demonstrating that NKG2D is an effective biomarker for MAGT1 correction. Despite the role of MAGT1 in homeostasis of intracellular Mg2+, magnesium supplements have not been effective for treatment. Potentially curative hematopoietic stem cell transplantation is associated with high mortality rates,4,13-15 and there is currently no gene therapy option for this newly described PID.
We sought to develop a gene therapy approach that would address most of the mutations and maintain endogenous regulation to ensure physiological expression using the CRISPR/Cas9 approach to target insertion (TI) of a corrective MAGT1 complementary DNA (cDNA) near the transcription start site.16-19 Exciting developments in CRISPR gene correction for several PIDs20-25 are unfortunately not matched by the pace of clinical translation.26 A major barrier to clinical translation of promising CRISPR gene correction procedures is the low engraftment rates of edited hematopoietic stem and progenitor cells (HSPCs) with substantially reduced correction rates after transplantation in mouse xenograft models, which indicates poor targeting of engrafting human HSPCs.27-30 We address this by transient suppression of both TP53-binding protein 1 (53BP1) and the p53 pathway to enhance the homology-directed repair (HDR) process and dampen DNA damage response (DDR), respectively, to achieve highly efficient MAGT1 correction in engrafting XMEN patient HSPCs for potential gene therapy. This versatile approach was also readily applied to correction of XMEN T cells, offering an alternate cell therapy option for XMEN disease.
Methods
Human samples
Healthy donors (HDs) and XMEN patients underwent apheresis after providing written informed consent (National Institute of Allergy and Infectious Diseases [NIAID] protocols 05-I-0213 and 94-I-0073) for collection of CD34+ HSPCs and peripheral blood mononuclear cells (PBMCs).
Recombinant adeno-associated virus serotype 6 (rAAV6)-MAGT1 donor template
The codon-optimized MAGT1 cDNA (Integrated DNA Technologies) was cloned into a pUC57 plasmid containing AAV vector genome inverted terminal repeats for production of rAAV6-MAGT1 donor template (Vigene Biosciences, Inc) for HDR.
Gene editing (GE) of T cells and HSPCs
Prestimulated PBMCs (as a source of T cells)24 or HSPCs31 were electroporated in the presence of glycerol, single-guide RNA (sgRNA), Cas9 from Streptococcus pyogenes, inhibitor of 53BP1 (i53), and human genetic suppressor element (hGSE) of p53 pathway mRNAs and then immediately transduced with rAAV6-MAGT1.
Transplantation studies
Triple transgenic immunodeficient NOD.Cg-PrkdcscidIl2rgtm1WjlTg (NSGS) mice expressing human interleukin-3, granulocyte-macrophage colony-stimulating factor, and stem cell factor cytokines32 (013062; The Jackson Laboratory) were used for transplantation studies (NIAID Institutional Animal Care and Use Committee approved protocol Laboratory of Clinical Immunology and Microbiology 1E [LCIM-1E]). Newborn mice (0-4 days old) that had been irradiated at 100 cGy were injected intrahepatically with 1 × 106 to 1.5 × 106 HSPCs per mouse. At 16 weeks, peripheral blood (PB), bone marrow (BM), spleen, and thymus were collected for analysis.
Flow cytometry
Flow cytometric analysis was performed using a BD Canto flow cytometer, DIVA software (BD Biosciences), and FlowJo 10.6.1 software (Tree Star) and by using monoclonal antibodies or apoptosis and proliferation kits as listed in supplemental Table 1 (available on the Blood Web site).
Molecular analysis
Genomic DNA was extracted using QIAamp DNA mini kit (QIAGEN). Quantification of indels at MAGT1 exon 1 was performed using tracking of indels by decomposition (TIDE) analysis33 after polymerase chain reaction (PCR) amplification and Sanger sequencing. Targeted insertion of MAGT1 cDNA was quantified by droplet digital PCR. Experimental identification of off-target (OT) sites was performed in vitro using circularization for high-throughput analysis of nuclease genome-wide effects by sequencing (CHANGE-seq).34
Statistical analysis
Differences between the means were tested using a Student t test or 1-way analysis of variance (ANOVA) with Tukey’s post hoc multiple comparisons test. Spearman’s correlation test was used to analyze the association between parameters. Statistical analyses were performed using GraphPad Prism (version 8.1.0) and statistical significance was indicated by providing P values.
Results
Optimization of HDR efficiency in CD34+ HSPCs
Of the 10 sgRNAs designed for CRISPR/Cas9 targeting of the MAGT1 gene transcription start site (Figure 1A), 5 achieved >70% cutting efficiency (Figure 1B) and sgRNA#1 was selected for the remaining studies because of its proximity to the ATG start codon (6 bp downstream) and a predicted highly specific profile (supplemental Table 2). Cas9 mRNA and sgRNA were delivered by electroporation into PB CD34+ HSPCs after a 2-day prestimulation post thaw. We compared the effects of varying amounts of Cas9 mRNA and glycerol on correction efficiency to identify optimal concentrations (25 μg/mL Cas9 mRNA, 2% glycerol; supplemental Figure 1A-B). We designed a corrective rAAV6-MAGT1 donor template for HDR containing an ∼3-kb codon-optimized MAGT1 cDNA, woodchuck hepatitis virus posttranscriptional regulatory element sequence to improve mRNA stability and maximize protein expression, β-globin polyadenylation signal, and homology arms complementary to the sequences adjoining the predicted site of the Cas9-induced double-strand break (DSB) at the target site (Figure 1C).
![Design of rAAV6-MAGT1 donor for CRISPR/Cas9 targeted integration into XMEN CD34+ HSPCs. (A) Location of the 10-candidate sgRNAs targeting MAGT1 gene transcription start site. Each sgRNA (sg) is represented as a 20-bp sequence (light pink or green) associated with the 3-bp protospacer adjacent motif sequence (darker color). (B) Evaluation of the percentage of cutting activity by tracking of indels by decomposition (TIDE) assay of indel formation after guide DNA (gDNA) extraction from untreated vs Cas9/sgRNA-treated HD CD34+ cells at 5 days post-electroporation (EP), sequencing of exon 1, and comparison of the sequences using TIDE software (data represent 1-5 independent experiments ). (C) Design of the rAAV6-MAGT1 donor. (D) Molecular analysis showing the percentage of TIs in gene-edited CD34+ cells (3 different donors, 2 HDs and XMEN patient 1) using GE enhancers as described. gDNA was extracted at 5 days post-EP, and insertion of the MAGT1 cDNA donor was quantified by droplet digital PCR (ddPCR) (4 independent experiments for –i53, 7 for +i53, and 7 for +i53+hGSE). (E) Viability was determined by trypan blue exclusion in naive and rAAV6-MAGT1–treated HSPCs at day 2 post-EP (5 independent experiments for –i53, 10 for +i53, and 10 for +i53+hGSE). (F) Bar graphs of HSPC subpopulations (multilymphoid progenitor [MLP], CD34+CD38+CD45RA–; common myeloid progenitor [CMP], CD34+CD38+CD45RA–; HSCs, CD34+CD38–CD45RA–CD90+CD133+; and multipotential progenitor [MPP], CD34+CD38–CD45RA–CD90–]) 2 days EP (data represent 4 independent experiments for –i53, 4 for +i53, and 7 for +i53+hGSE), compared with naive cells. Data are shown as mean ± standard deviation (SD); analysis of variance (ANOVA) 1-way test and Tukey’s post hoc multiple comparisons test were used. *P < .05; **P < .01; ***P < .001; ****P < .0001. Fwd, forward; ITR, inverted terminal repeat; ns, nonsignificant; LHA, left homology arm; polyA, poly(A) tail; RHA, right homology arm; Rev, reverse; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/26/10.1182_blood.2021011192/4/m_bloodbld2021011192f1.png?Expires=1769295971&Signature=wyoRYOy1bM8wZHoGu7BlFxhXwpuUHcr7U-W4tHZmFVXCmjXVuNiu4HXMWON8Ko8-vWpsFpVe1H4M9Hdvt7pluZGw0p66IK195Y2DeqhsDRZp1i~ZOf7xY5w00bvHiizi4o3RwHHnxW21Amq~U1OCEzorB9j7jST3PjP6TR~l3rJblyLZmyjYx0JmAAT16J0li5Cs74vQLuJScYmiEwnlGGGsWwaZjEGEO0cYMAyHRpEO23G~RAqep~DSsT8W~oXarraq2EYul0bo31~-16zMZSoFRfQgyxZTzLMIKDwtU2hbeu9rg1qivFaEx0tBh1FEY87x0Ss3BLTcGnbPbHACZQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Design of rAAV6-MAGT1 donor for CRISPR/Cas9 targeted integration into XMEN CD34+ HSPCs. (A) Location of the 10-candidate sgRNAs targeting MAGT1 gene transcription start site. Each sgRNA (sg) is represented as a 20-bp sequence (light pink or green) associated with the 3-bp protospacer adjacent motif sequence (darker color). (B) Evaluation of the percentage of cutting activity by tracking of indels by decomposition (TIDE) assay of indel formation after guide DNA (gDNA) extraction from untreated vs Cas9/sgRNA-treated HD CD34+ cells at 5 days post-electroporation (EP), sequencing of exon 1, and comparison of the sequences using TIDE software (data represent 1-5 independent experiments ). (C) Design of the rAAV6-MAGT1 donor. (D) Molecular analysis showing the percentage of TIs in gene-edited CD34+ cells (3 different donors, 2 HDs and XMEN patient 1) using GE enhancers as described. gDNA was extracted at 5 days post-EP, and insertion of the MAGT1 cDNA donor was quantified by droplet digital PCR (ddPCR) (4 independent experiments for –i53, 7 for +i53, and 7 for +i53+hGSE). (E) Viability was determined by trypan blue exclusion in naive and rAAV6-MAGT1–treated HSPCs at day 2 post-EP (5 independent experiments for –i53, 10 for +i53, and 10 for +i53+hGSE). (F) Bar graphs of HSPC subpopulations (multilymphoid progenitor [MLP], CD34+CD38+CD45RA–; common myeloid progenitor [CMP], CD34+CD38+CD45RA–; HSCs, CD34+CD38–CD45RA–CD90+CD133+; and multipotential progenitor [MPP], CD34+CD38–CD45RA–CD90–]) 2 days EP (data represent 4 independent experiments for –i53, 4 for +i53, and 7 for +i53+hGSE), compared with naive cells. Data are shown as mean ± standard deviation (SD); analysis of variance (ANOVA) 1-way test and Tukey’s post hoc multiple comparisons test were used. *P < .05; **P < .01; ***P < .001; ****P < .0001. Fwd, forward; ITR, inverted terminal repeat; ns, nonsignificant; LHA, left homology arm; polyA, poly(A) tail; RHA, right homology arm; Rev, reverse; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element.
Design of rAAV6-MAGT1 donor for CRISPR/Cas9 targeted integration into XMEN CD34+ HSPCs. (A) Location of the 10-candidate sgRNAs targeting MAGT1 gene transcription start site. Each sgRNA (sg) is represented as a 20-bp sequence (light pink or green) associated with the 3-bp protospacer adjacent motif sequence (darker color). (B) Evaluation of the percentage of cutting activity by tracking of indels by decomposition (TIDE) assay of indel formation after guide DNA (gDNA) extraction from untreated vs Cas9/sgRNA-treated HD CD34+ cells at 5 days post-electroporation (EP), sequencing of exon 1, and comparison of the sequences using TIDE software (data represent 1-5 independent experiments ). (C) Design of the rAAV6-MAGT1 donor. (D) Molecular analysis showing the percentage of TIs in gene-edited CD34+ cells (3 different donors, 2 HDs and XMEN patient 1) using GE enhancers as described. gDNA was extracted at 5 days post-EP, and insertion of the MAGT1 cDNA donor was quantified by droplet digital PCR (ddPCR) (4 independent experiments for –i53, 7 for +i53, and 7 for +i53+hGSE). (E) Viability was determined by trypan blue exclusion in naive and rAAV6-MAGT1–treated HSPCs at day 2 post-EP (5 independent experiments for –i53, 10 for +i53, and 10 for +i53+hGSE). (F) Bar graphs of HSPC subpopulations (multilymphoid progenitor [MLP], CD34+CD38+CD45RA–; common myeloid progenitor [CMP], CD34+CD38+CD45RA–; HSCs, CD34+CD38–CD45RA–CD90+CD133+; and multipotential progenitor [MPP], CD34+CD38–CD45RA–CD90–]) 2 days EP (data represent 4 independent experiments for –i53, 4 for +i53, and 7 for +i53+hGSE), compared with naive cells. Data are shown as mean ± standard deviation (SD); analysis of variance (ANOVA) 1-way test and Tukey’s post hoc multiple comparisons test were used. *P < .05; **P < .01; ***P < .001; ****P < .0001. Fwd, forward; ITR, inverted terminal repeat; ns, nonsignificant; LHA, left homology arm; polyA, poly(A) tail; RHA, right homology arm; Rev, reverse; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element.
Homology-directed repair of the DSB is necessary to incorporate the donor sequence, but it is limited to the S/G2 phase of the cell cycle and competes with the alternate canonical repair pathway by nonhomologous end joining (NHEJ) that is rapid, dominant, and active in all cell cycle phases.35 We transiently blocked NHEJ and skewed the competition to favor HDR by transient inhibition of 53BP1 with i53 to block 53BP1 accumulation at the DSB site that is essential for NHEJ repair.36,37 Increased TI rates from 31.45% ± 15.1% to 50.0% ± 18.2% with i53 confirmed its effectiveness at promoting HDR-mediated TI in HSPCs (Figure 1D). However, we observed a loss in cell viability compared with the untreated (naive) cells at 2 days after editing, suggesting toxicities unrelated to i53 (Figure 1E).
Next, we assessed the impact of editing on the stemness of HSPCs by determining the relative proportions of CD34+ subpopulations. Cells treated with AAV showed an increase in differentiation markers with increased proportions of common myeloid progenitor (CD34+CD38+CD45RA–) and decreased hematopoietic stem cell (HSC) and multipotential progenitor (CD34+CD38–CD45RA–) subpopulations (Figure 1F; supplemental Figure 1C).
DNA damage response to AAV transduction
To further investigate these concerns of cytotoxicity related to GE, we assessed phosphorylation of H2AX (γH2AX) as a marker of DDR. Exposure to AAV alone, even in the absence of Cas9/sgRNA-induced DSBs, elicited a sharp increase in percentages of γH2AX+ cells observable by 2 days (Figure 2A) to 24 hours (Figure 2B-C), which was indicative of a massive early-onset DDR upon exposure that decreased significantly by 48 hours after treatment (Figure 2C). DDR pathways are controlled by ataxia telangiectasia mutated (ATM), ATM- and Rad3-related (ATR), and DNA-dependent protein kinase which, upon activation, phosphorylate various proteins, including H2AX, to orchestrate DNA repair and maintain genomic integrity.38 We evaluated the effects of caffeine (4 mM), a known inhibitor of ATM/ATR, on HSPC GE during AAV transduction. Caffeine decreased H2AX phosphorylation by 40% at 24 hours (Figure 2B), confirming AAV-induced activation of ATM/ATR kinases.39,40
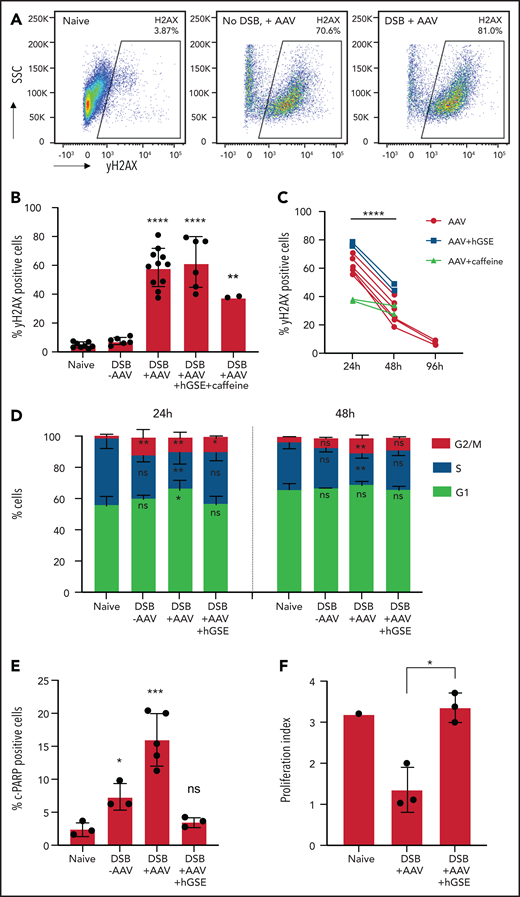
Effect of hGSE mRNA on DDR after GE with AAV. (A) Representative flow cytometric dot plots showing γH2AX expression in CD34+ cells (naive, AAV alone without Cas9/sgRNA-induced DSB, Cas9/sgRNA-induced DSB + AAV) 2 hours after treatment. (B) Histogram summarizing the percentages of γH2AX+ marker in the conditions indicated at 24 hours (h) after DSB induced by Cas9/sgRNA (data represent 2-7 independent experiments). (C) Evolution of percentages of γH2AX+ cells after 48 to 96 hours after treatment (data represent 2-7 independent experiments). Paired sample Student t test was used to compare the proportion of positive cells at 24 and 48 hours. (D) Percentage of CD34+ cells in G1, S, or G2/M cell cycle phases at 24 and 48 hours after treatment. Significance compared with naive condition is indicated. (E) Histograms summarizing the percentages of cleaved poly(ADP-ribose) polymerase (c-PARP)+ cells, a marker of apoptosis, at 24 hours in the conditions indicated (data are representative of 3 independent experiments). (F) Proliferation index (FlowJo software) comparing carboxyfluorescein succinimidyl ester (CFSE)–stained HSPCs at day 1 and day 6 post-EP (data are representative of 3 independent experiments). Data are shown as mean ± SD. ANOVA 1-way test and Tukey’s post hoc multiple comparisons test were used. *P < .05; **P < .01; ***P < .001; ****P < .0001. SSC, side scatter.
Effect of hGSE mRNA on DDR after GE with AAV. (A) Representative flow cytometric dot plots showing γH2AX expression in CD34+ cells (naive, AAV alone without Cas9/sgRNA-induced DSB, Cas9/sgRNA-induced DSB + AAV) 2 hours after treatment. (B) Histogram summarizing the percentages of γH2AX+ marker in the conditions indicated at 24 hours (h) after DSB induced by Cas9/sgRNA (data represent 2-7 independent experiments). (C) Evolution of percentages of γH2AX+ cells after 48 to 96 hours after treatment (data represent 2-7 independent experiments). Paired sample Student t test was used to compare the proportion of positive cells at 24 and 48 hours. (D) Percentage of CD34+ cells in G1, S, or G2/M cell cycle phases at 24 and 48 hours after treatment. Significance compared with naive condition is indicated. (E) Histograms summarizing the percentages of cleaved poly(ADP-ribose) polymerase (c-PARP)+ cells, a marker of apoptosis, at 24 hours in the conditions indicated (data are representative of 3 independent experiments). (F) Proliferation index (FlowJo software) comparing carboxyfluorescein succinimidyl ester (CFSE)–stained HSPCs at day 1 and day 6 post-EP (data are representative of 3 independent experiments). Data are shown as mean ± SD. ANOVA 1-way test and Tukey’s post hoc multiple comparisons test were used. *P < .05; **P < .01; ***P < .001; ****P < .0001. SSC, side scatter.
Previous studies reported the detrimental effects of the DDR-activated p53 pathway after nuclease-induced DSBs, including cell cycle arrest, senescence, or apoptosis,41-43 which could be alleviated by adding a rat-derived TP53 inhibitor or a genetic suppressor element (GSE).41 To avoid potential immune responses to rat-derived sequences, we evaluated the use of a humanized TP53 inhibitor/hGSE mRNA, which maintained TI rates (62.3% ± 8%) (Figure 1D) with significantly improved cell viability (Figure 1E) and preserved HSC subpopulations at a level comparable to that of naive cells (Figure 1F). A colony-forming unit assay also demonstrated that HSPCs edited in the presence of hGSE retained their hematopoietic potential (supplemental Figure 1D).
We also performed cell cycle analysis which revealed that at 24 hours after editing, there was significant G1 arrest exacerbated by AAV that was mitigated with hGSE, which led to an increase in G2/M phase cells and a relative preservation of S phase in the presence of hGSE (Figure 2D). By 48 hours after editing, the distribution of cells in different phases had normalized except for cells edited with AAV without hGSE. We also assessed cell apoptosis that resulted from DDR by evaluating cleaved poly(ADP-ribose) polymerase 1 (c-PARP-1; a biomarker of apoptosis) expression, demonstrating a significant decrease in apoptosis at 24 hours after GE (Figure 2E) and preservation of proliferative capacity (Figure 2F; supplemental Figure 1E) with transient suppression of p53. Taken together, these data confirm the effects of hGSE in mitigating adverse responses to AAV for GE.
In vitro phenotypic and functional correction in T and NK cells differentiated from gene edited XMEN HSPCs
Because MAGT1-dependent expression of NKG2D is restricted to mature cells (supplemental Figure 2A), we assessed rescue of functional MAGT1 correction by evaluating NKG2D expression after in vitro differentiation of edited HSPCs. With the use of an artificial thymic organoid (ATO) system that was recently instrumental in characterizing T-cell differentiation defects in PIDs,44,45 edited XMEN HSPCs underwent T-cell differentiation and expressed NKG2D in CD3+ T cells that was indistinguishable from that of HD cells (Figure 3A; supplemental Figure 2B). Molecular analysis of cells harvested from the ATOs confirmed maintenance of TI in the MAGT1 gene (Figure 3B). In addition, using a K562 erythroleukemia cell line modified for constitutive surface expression of supportive cytokine interleukin 15 and stimulatory 4-1BB ligand,46 we efficiently differentiated edited XMEN HSPCs in vitro into NK cells (CD3–CD56+) (Figure 3C). The percentage of NKG2D-expressing NK cells almost doubled with i53, and it was highest with i53 and hGSE combined (–i53, 15.9% ± 8.1%; +i53, 29.2% ± 15.9%; +i53+hGSE, 35% ± 16.1%) (Figure 3D), and the level of expression was also similar to that of HD NK cells (Figure 3E). Molecular droplet digital PCR assessment confirmed significantly improved TI rates with the combined enhancers (47.0% ± 15.5% with i53+hGSE vs 26.6% ± 16.9% without enhancers) (Figure 3F). Furthermore, we found that NK cytotoxicity was comparable between edited XMEN and HD cells at an effector:target ratio of 2:1 (Figure 3G; supplemental Figure 2C), and that the killing activity correlated with the level of NKG2D expression (Figure 3H). Thus, our data demonstrated that gene-edited XMEN HSPCs maintained their capability to differentiate to T and NK cells, with restored MAGT1 glycosylation function that led to the expression of NKG2D in T and NK cells.

In vitro phenotypic and functional correction in immune cells after GE of XMEN CD34+ HSPCs. (A) Dot plots showing the gating strategy after in vitro T-cell differentiation of CD34+ cells using the ATO system; bar graph on the right shows NKG2D expression in CD3+ T cells at 6 weeks of differentiation (data are representative of 2-4 independent experiments; 300-20 000 events acquired in CD3+ gate). (B) Targeted integration measured by ddPCR analysis in ATO-derived cells at week 6 of in vitro differentiation (data are representative of 2 independent experiments). (C) Dot plot showing the gating for NK cells (CD3–CD56+) and NKG2D expression after 35 days of in vitro NK-cell differentiation from CD34+ cells; bar graph shows the percentage of CD34-derived NK cells in each condition. (D) NKG2D expression (% positive cells), (E) level of expression determined as the percentage of mean fluorescence intensity (MFI) of HD NK cells, and (F) targeted integration by ddPCR analysis in NK cells at day 35 of in vitro differentiation (4 independent experiments for –i53, 5 for +i53, and 8 for +i53+hGSE). (G) Cytotoxic activity of CD34+-differentiated NK cells against K562 cells at an effector:target ratio of 2:1 (4 independent experiments for –i53, 9 for +i53, and 8 for +i53+hGSE). (H) Correlation between the NKG2D expression (%) and the killing activity (%) in NK cells at day 35 was calculated using Spearman’s correlation coefficient and 2-tailed P value (n = 33 pairs). Data are shown as mean ± SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. FSC, forward scatter.
In vitro phenotypic and functional correction in immune cells after GE of XMEN CD34+ HSPCs. (A) Dot plots showing the gating strategy after in vitro T-cell differentiation of CD34+ cells using the ATO system; bar graph on the right shows NKG2D expression in CD3+ T cells at 6 weeks of differentiation (data are representative of 2-4 independent experiments; 300-20 000 events acquired in CD3+ gate). (B) Targeted integration measured by ddPCR analysis in ATO-derived cells at week 6 of in vitro differentiation (data are representative of 2 independent experiments). (C) Dot plot showing the gating for NK cells (CD3–CD56+) and NKG2D expression after 35 days of in vitro NK-cell differentiation from CD34+ cells; bar graph shows the percentage of CD34-derived NK cells in each condition. (D) NKG2D expression (% positive cells), (E) level of expression determined as the percentage of mean fluorescence intensity (MFI) of HD NK cells, and (F) targeted integration by ddPCR analysis in NK cells at day 35 of in vitro differentiation (4 independent experiments for –i53, 5 for +i53, and 8 for +i53+hGSE). (G) Cytotoxic activity of CD34+-differentiated NK cells against K562 cells at an effector:target ratio of 2:1 (4 independent experiments for –i53, 9 for +i53, and 8 for +i53+hGSE). (H) Correlation between the NKG2D expression (%) and the killing activity (%) in NK cells at day 35 was calculated using Spearman’s correlation coefficient and 2-tailed P value (n = 33 pairs). Data are shown as mean ± SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. FSC, forward scatter.
Engraftment of gene-edited XMEN HSPCs and in vivo correction
A major determining factor for clinical translation of GE is the durability of the gene correction, which is dependent on the correction of true engrafting HSPCs. To assess this, we transplanted gene-edited XMEN HSPCs into irradiated NSGS newborn mouse pups, a model that allows the development of lymphocytes in vivo.32 The engraftment (human CD45+) rates of XMEN HSPCs edited in the presence of enhancers were comparable with those of HD cells in mouse BM (HD, 16.4% ± 12.1%; XMEN naive, 5.5% ± 2.2%; gene-edited XMEN, 2.8% ± 1.2% without enhancers, 11.2% ± 9.1% with i53, and 11.8% ± 8.9% with i53+hGSE), with successful colonization of multiple organs (PB, spleen, and thymus) at 16 weeks after transplant (Figure 4A). The percentages of myeloid cells (CD33+), B cells (CD19+), and T cells (CD3+) in BM was comparable to that of HD controls, indicating that differentiation capability was maintained in gene-edited XMEN HSPCs (Figure 4B). In addition, low levels of human CD34+ cells remained detectable in BM (Figure 4B).
![Hematopoietic reconstitution of gene-edited HSPCs in immunodeficient mice. (A) Human engraftment measured by the presence of human CD45+ cells by flow cytometry at week 16 in the BM, PB, spleen, and thymus of NSGS mice transplanted with HD, naive (n = 15 mice), or gene-edited XMEN CD34+ HSPCs (n = 3 mice [–i53, 2 experiments], 12 mice [+i53, 5 experiments], and 24 mice [+i53+hGSE, 4 experiments]). The dotted line indicates the threshold for engraftment at 0.2% hCD45+ cells in the BM. (B) Immunophenotypic analysis of BM at 16 weeks posttransplant by flow cytometry showing the percentages of CD34+ cells, myeloid (CD33+), lymphoid B (CD19+), and T (CD3+) after gating on the hCD45+ population. (C-D) Percentage of NKG2D expression in NK cells from the spleen (C) and CD8+ T cells from the PB (D), spleen, and thymus at week 16 posttransplant. (E) Targeted insertion quantified by ddPCR in human CD45+ cells isolated from the BM after GE without enhancers (red bar), with i53 (blue bar), or with i53+hGSE (green bar); BM from the mice injected with the same cells were pooled. (F) Targeted insertion quantified by ddPCR in the lymphoid T (CD3), myeloid (CD33/CD14), and lymphoid B (CD19) cells sorted from the spleen after GE with i53+hGSE. Data are shown as mean ± SD.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/26/10.1182_blood.2021011192/4/m_bloodbld2021011192f4.png?Expires=1769295971&Signature=SzEdv91cUmabuE0e5oRPjoSBQsveafpRk-nsVYyGOhmo4PGMJLdGADYyotR61rVG4ZAk2HJn~r-huQELeuPZg1D0r9PBfS7aL3QeKUuW57KDRTqINFTRhwTfrTmgTWYfto43iBzlVSqaKsfoSNao~tPWlrIUkik1HUmC9MnIdlFZd4XGg462~SMKvlefwcoOrj7K4MIXSlr2lT1ZqDRC6pXddBNklYNuROU7X~nFO~8OCRsRGfTLqT-f2MwEroe2SyJcawklU7Wf9OxSto7UCM0TtTsRWa9DjhGIhfmsl5SlO-fe8cAHyijeCtFjjzdU2AhiYjIm5mLtqfq0ZHooVw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Hematopoietic reconstitution of gene-edited HSPCs in immunodeficient mice. (A) Human engraftment measured by the presence of human CD45+ cells by flow cytometry at week 16 in the BM, PB, spleen, and thymus of NSGS mice transplanted with HD, naive (n = 15 mice), or gene-edited XMEN CD34+ HSPCs (n = 3 mice [–i53, 2 experiments], 12 mice [+i53, 5 experiments], and 24 mice [+i53+hGSE, 4 experiments]). The dotted line indicates the threshold for engraftment at 0.2% hCD45+ cells in the BM. (B) Immunophenotypic analysis of BM at 16 weeks posttransplant by flow cytometry showing the percentages of CD34+ cells, myeloid (CD33+), lymphoid B (CD19+), and T (CD3+) after gating on the hCD45+ population. (C-D) Percentage of NKG2D expression in NK cells from the spleen (C) and CD8+ T cells from the PB (D), spleen, and thymus at week 16 posttransplant. (E) Targeted insertion quantified by ddPCR in human CD45+ cells isolated from the BM after GE without enhancers (red bar), with i53 (blue bar), or with i53+hGSE (green bar); BM from the mice injected with the same cells were pooled. (F) Targeted insertion quantified by ddPCR in the lymphoid T (CD3), myeloid (CD33/CD14), and lymphoid B (CD19) cells sorted from the spleen after GE with i53+hGSE. Data are shown as mean ± SD.
Hematopoietic reconstitution of gene-edited HSPCs in immunodeficient mice. (A) Human engraftment measured by the presence of human CD45+ cells by flow cytometry at week 16 in the BM, PB, spleen, and thymus of NSGS mice transplanted with HD, naive (n = 15 mice), or gene-edited XMEN CD34+ HSPCs (n = 3 mice [–i53, 2 experiments], 12 mice [+i53, 5 experiments], and 24 mice [+i53+hGSE, 4 experiments]). The dotted line indicates the threshold for engraftment at 0.2% hCD45+ cells in the BM. (B) Immunophenotypic analysis of BM at 16 weeks posttransplant by flow cytometry showing the percentages of CD34+ cells, myeloid (CD33+), lymphoid B (CD19+), and T (CD3+) after gating on the hCD45+ population. (C-D) Percentage of NKG2D expression in NK cells from the spleen (C) and CD8+ T cells from the PB (D), spleen, and thymus at week 16 posttransplant. (E) Targeted insertion quantified by ddPCR in human CD45+ cells isolated from the BM after GE without enhancers (red bar), with i53 (blue bar), or with i53+hGSE (green bar); BM from the mice injected with the same cells were pooled. (F) Targeted insertion quantified by ddPCR in the lymphoid T (CD3), myeloid (CD33/CD14), and lymphoid B (CD19) cells sorted from the spleen after GE with i53+hGSE. Data are shown as mean ± SD.
Analysis of mouse PB, spleen, and thymus also confirmed robust human T- and NK-cell development (supplemental Figure 3A-B), independent of enhancers (i53, hGSE) and normal differentiation to mature CD4+, CD8+, and double-positive T cells (supplemental Figure 3C). Restoring specific NKG2D expression in XMEN edited NK and CD8+ T cells to the level of HD cells confirmed functional correction of MAGT1 (Figure 4C-D).
Importantly, molecular analysis in BM-sorted human CD45+(hCD45+) cells revealed a remarkable persistence of enhanced TI rates at 63.1% ± 8.8% compared with in vitro TI rates (62.3% ± 8%; Figure 1D) when edited with i53+hGSE or with i53 alone (54.5% ± 18.3% in vivo vs 50.0% ± 18.2% in vitro) (Figure 4E), indicating efficient targeting of engrafting HSPCs. TI was also confirmed in human myeloid cells, lymphoid B cells, and spleen T cells (Figure 4F). Overall, we demonstrated efficient GE of engraftable XMEN HSPCs that successfully differentiated into NKG2D-expressing NK and CD8+ T cells in vivo.
Efficient gene correction of apheresis XMEN T cells
Defective lymphocytes account for the major morbidity rates in XMEN patients with chronic EBV infections and lymphoproliferative disease. In addition, there is a latency for competent T-cell development after successful HSC transplant. We have previously shown that correction of T cells with MAGT1 mRNA restores NKG2D expression and lymphocyte function.3,12 Here we sought to genetically correct XMEN lymphocytes using the approach optimized in HSPCs (Cas9/sgRNA/AAV). At 2 days after editing, we observed restoration of MAGT1 expression (Figure 5A) and a range of 4% to 42% of NKG2D-expressing XMEN CD8+ T cells (Figure 5B) that correlated with TI percentages (Figure 5C), both of which increased significantly with time in culture (Figure 5B-C). In addition, the level of restored NKG2D expression in corrected XMEN T cells also reached the same level as that in HD T cells (Figure 5D), as did CD70, another MAGT1 glycosylated protein, but not CD28 (Figure 5E). These data confirmed that MAGT1 correction in T cells restores the glycosylation of proteins such as NKG2D and CD70. The growth advantage of gene-corrected cells during in vitro culture indicates a favorable potential for cell therapy with gene-corrected T cells for short-term treatment of infections in XMEN patients.
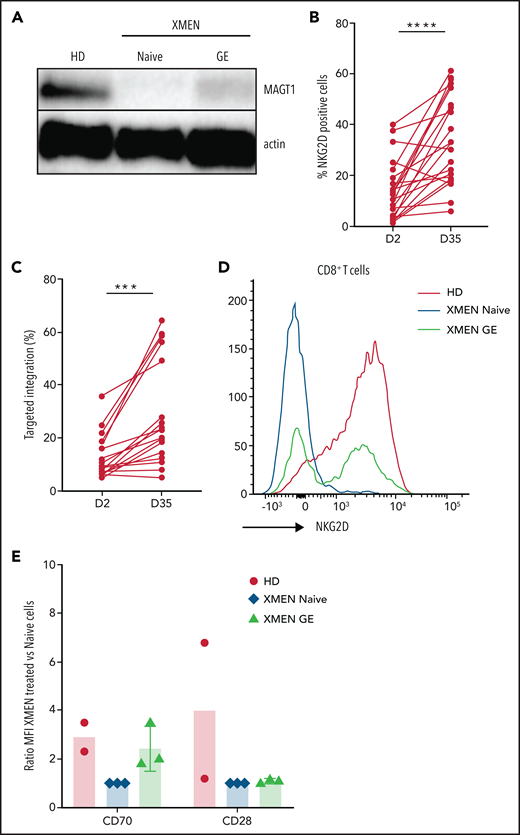
GE in XMEN T cells. (A) Representative western blot for MAGT1 protein expression in HD XMEN (naive and gene edited) T cells with β-actin as loading control. (B) Percentage of NKG2D-positive CD8+ T cells in AAV-treated T cells at days 2 and 35 post-EP (data are representative of 8 independent experiments; XMEN patients P1 and P2). (C) Targeted insertion rates measured at day 2 and day 35 post-EP (data are representative of 8 independent experiments). (D) Representative flow cytometry histogram overlaying NKG2D expression in HD and XMEN (naive and gene-edited) T cells at day 35 after GE. Percentage of NKG2D+ in gene-edited CD8+ XMEN T cells was 57.5% and TI was 64.6%. (E) Expression of CD70 and CD28 by flow cytometry for HD and XMEN (naive and gene-edited) T cells at day 28 post-EP; data are expressed as the ratio of MFI normalized to XMEN untreated cells for CD70 and CD28 (data are representative of 2-3 independent experiments). A paired sample Student t test was used. ***P < .001; ****P < .0001.
GE in XMEN T cells. (A) Representative western blot for MAGT1 protein expression in HD XMEN (naive and gene edited) T cells with β-actin as loading control. (B) Percentage of NKG2D-positive CD8+ T cells in AAV-treated T cells at days 2 and 35 post-EP (data are representative of 8 independent experiments; XMEN patients P1 and P2). (C) Targeted insertion rates measured at day 2 and day 35 post-EP (data are representative of 8 independent experiments). (D) Representative flow cytometry histogram overlaying NKG2D expression in HD and XMEN (naive and gene-edited) T cells at day 35 after GE. Percentage of NKG2D+ in gene-edited CD8+ XMEN T cells was 57.5% and TI was 64.6%. (E) Expression of CD70 and CD28 by flow cytometry for HD and XMEN (naive and gene-edited) T cells at day 28 post-EP; data are expressed as the ratio of MFI normalized to XMEN untreated cells for CD70 and CD28 (data are representative of 2-3 independent experiments). A paired sample Student t test was used. ***P < .001; ****P < .0001.
Identification and quantification of OT events
Candidate OT sites were identified using an ultrasensitive, unbiased, genome-wide in vitro CHANGE-seq approach.34 As shown in the Manhattan plot, we observed robust editing of the on-target site and some evidence of in vitro targeting of several OT events (Figure 6A), with estimated specificity ratios of 0.14 to 0.36 (Figure 6B). Most of these OT event sites were located in intergenic (36% to 40%) and gene intronic (40% to 46%) regions, whereas 14% to 20% were located in exonic regions (Figure 6C). To verify potential indels at predicted sites, we performed high-throughput sequencing of the top 8 OT event candidates (Figure 6D; supplemental Table 3) and observed no increase in indels in edited (in vitro pretransplant and in vivo posttransplant) samples compared with untreated naive samples (Figure 6E). Reassuringly, the addition of i53+hGSE did not increase the frequency of OT indels.
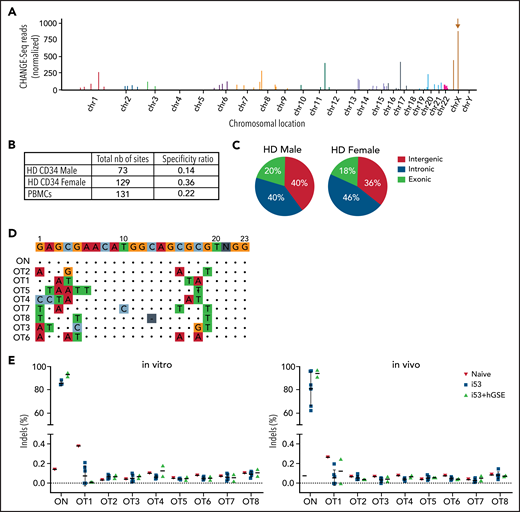
Analysis of OT activity. (A) Manhattan plot showing the results of off-targets for CD34+ cells by CHANGE-seq assay using gDNA extracted from HD male donor CD34+ cells electroporated with Cas9 mRNA and sgRNA#1. The brown arrow indicates the on-target site (MAGT1 gene) on chromosome X. (B) Table reporting the total number (nb) of cleavage sites (on- and off-targets) and the specificity ratio using male and female HD HSPCs or HD peripheral blood mononuclear cells (PBMCs). (C) Pie charts showing fraction of cleavage sites categorized according to their genomic features after CD34+ cells from HD male and female donors were edited. (D) Identity of the nucleotide mismatches at the OT sites. ON indicates the on-target site without mismatch, whereas OT1-OT8 indicate the top 8 OT sites shared by male and female HD CD34+ HSPCs and HD PBMCs. (E) Percentage of cutting activity evaluated by sequencing at ON and OT sites detected by CHANGE-Seq in in vitro (CD34+ cells 2 days after gene editing) and in vivo (hCD45+ cells from the BM of NSGS mice that had received a transplant at week 16 posttransplant) samples. For all the graphs, data are shown as mean ± SD.
Analysis of OT activity. (A) Manhattan plot showing the results of off-targets for CD34+ cells by CHANGE-seq assay using gDNA extracted from HD male donor CD34+ cells electroporated with Cas9 mRNA and sgRNA#1. The brown arrow indicates the on-target site (MAGT1 gene) on chromosome X. (B) Table reporting the total number (nb) of cleavage sites (on- and off-targets) and the specificity ratio using male and female HD HSPCs or HD peripheral blood mononuclear cells (PBMCs). (C) Pie charts showing fraction of cleavage sites categorized according to their genomic features after CD34+ cells from HD male and female donors were edited. (D) Identity of the nucleotide mismatches at the OT sites. ON indicates the on-target site without mismatch, whereas OT1-OT8 indicate the top 8 OT sites shared by male and female HD CD34+ HSPCs and HD PBMCs. (E) Percentage of cutting activity evaluated by sequencing at ON and OT sites detected by CHANGE-Seq in in vitro (CD34+ cells 2 days after gene editing) and in vivo (hCD45+ cells from the BM of NSGS mice that had received a transplant at week 16 posttransplant) samples. For all the graphs, data are shown as mean ± SD.
Discussion
Great strides in the availability of whole-exome sequencing has led to the recent discovery of many PIDs such as XMEN disease. To address most of the pathogenic mutations that span MAGT1, we targeted the insertion of a MAGT1 cDNA donor template near the transcription start site for physiological regulation by the endogenous promoter. Despite promising levels of in vitro correction levels,27-30 failure to maintain GE rates in engrafted cells is a major impediment to clinical translation.27-30 Schiroli et al41 demonstrated significant DDR after genome editing of HSPCs with the widely used AAV vectors for donor template delivery.20,21,24,25,30,47 Our data on γH2AX expression kinetics confirm the DDR that occurs rapidly after exposure to AAV, even in the absence of nuclease-induced DSBs. This massive DDR causes widespread detrimental effects with induction of apoptosis, cell death, and cell cycle arrest that impairs cell fitness and engraftment capabilities that are demonstrable only after transplant. This DDR pertains particularly to AAV donors and significantly less so to oligonucleotide donors.37 By using a humanized version of the dominant negative TP53 (hGSE) to transiently dampen AAV-mediated DDR effects, we achieved robust engraftment of edited XMEN HSPCs with HDR enhanced by i53.
The precise mechanism for AAV triggering of DDR remains unclear. Our data demonstrating reduced γH2AX expression after treatment with caffeine are consistent with the involvement of ATR/ATM kinases in AAV-induced DDR as previously reported.39,48,49 Although the mechanism was initially attributed to the inverted terminal repeats flanking the AAV genome,50 Fragkos et al51 subsequently identified a cis-acting replication element in the p5 promoter of AAV2 DNA capable of initiating stalled replication complexes and stimulating DDR, and thus recombinant AAV is devoid of that activity. Our data showed the contrary because rAAV6-MAGT1 triggered robust DDR. Future research in AAV engineering to minimize DDR is important for improving the fitness of HSPCs gene edited with AAV donors. Our current approach using hGSE mRNA to transiently suppress the massive DDR after exposure to AAV promotes engraftment to achieve clinically relevant rates of correction after transplantation of gene-edited HSPCs.
Although gene correction of HSPCs can provide definitive treatment, human T-cell reconstitution from engrafted HSPCs can take a period of many months during which the patients are at increased risk of infections. Infections in patients with PIDs that provide the rationale for hematopoietic stem cell transplantation also significantly increase the risks associated with transplant myeloablation. Therefore, cellular therapy that provides immediate protection may offer an important therapeutic bridge or a short-term therapeutic option. The same CRISPR/Cas9 approach was easily applied to XMEN T cells, in which gene-edited XMEN T cells showed restored MAGT1 expression and N-glycosylation function, as indicated by the increased expression of N-glycoproteins NKG2D and CD70 at a physiological level which was, in the case of NKG2D, restricted to CD8+ T cells. Of interest, CD28 expression in the edited T cells did not increase significantly, consistent with our previous observation in XMEN T cells corrected with MAGT1 mRNA.12 This may be attributable to the expansion of residual CD28-expressing XMEN T cells by the CD3 and CD28 beads used in our cell culture proliferation.
Importantly, our data demonstrate a survival advantage in gene-corrected T lymphocytes with increased correction rates over time, suggesting the possibility of achieving clinical benefit from even low numbers of corrected cells. This survival advantage accounts for the outgrowth of normal cells in PB from XMEN female carriers with markedly skewed lyonization favoring the normal X-allele,1,5,8,15 with mutations detectable only in BM cells.8 CRISPR/Cas9 editing of chimeric antigen receptor T cells is currently being studied in a phase 1 trial for the treatment of refractory cancer.52 Additional preclinical studies with in vivo infusion of the gene-corrected T cells in immunodeficient mice will help evaluate the persistence of correction over time. Cellular therapy with gene-edited autologous lymphocytes for prompt control of EBV infections in XMEN patients before high-risk chemotherapy therefore offers an attractive therapeutic bridging option.
To address the limited human lymphocyte development typically observed in HSPC xenografts in adult NSG mice, we transplanted edited XMEN HSPCs into the liver of newborn pups20,25 of the NSGS mouse strain, which provides constitutive expression of human cytokines to support robust lymphocyte development.32 The efficient and stable in vivo gene correction achieved with our CRISPR/Cas9/AAV6 approach using gene-edited enhancers in XMEN HSPCs restored MAGT1 glycosylation function and expression of NKG2D receptor on NK and CD8+ T cells after HSPC transplant, with high TI rates (∼60%) maintained in engrafted NSGS mice. The percentages of NKG2D expression in CD8+ T cells from the spleen were slightly higher when treated with i53+hGSE (53% in blood, 61.2% in spleen, and 62.7% in thymus) than when treated with i53 only (41.8%, 56%, and 58.1%) despite similar engraftment rates, indicating a relative increase in correction of engrafting HSPCs when treated with both gene-edited enhancers.
Because functional correction of lymphocytes from spleen and thymus of transplanted mice could be assessed only several months after HSPC engraftment, we also demonstrated reliable tools for in vitro differentiation of T and NK cells using the recently described ATO system, which has been reported to support the development and differentiation of HSPCs into T lymphocytes44 and has allowed for the characterization of T-cell differentiation blockades that result from a variety of genetic causes.45 By using the ATO system, we showed that our gene editing process retains the capability of HSPCs to differentiate into CD3+ T cells, which allows us to assess the functional correction of MAGT1 glycosylation by measuring NKG2D expression. In parallel, we harnessed the capability of K562-IL15-41BBL stimulatory cells used for clinical large-scale expansion of NK cells46 supplemented with hematopoietic cytokines to establish an efficient in vitro NK differentiation protocol from the edited XMEN HSPCs. This allowed us to demonstrate corrected MAGT1 glycosylation function as well as NK cell cytolytic activity. These two in vitro models provide robust tools for assessing MAGT1 functional correction in 5 to 6 weeks.
For clinical application, safety considerations are as important as correction efficiency. A common concern for the use of CRISPR/Cas9 nucleases is the risk of OT DNA cuts that result in genomic insertions or deletions. There are evolving nucleases with improved specificities or nickases that avoid double-strand DNA cuts,53-57 but the accompanying decrease in editing efficiency must be considered. OT risks are also highly dependent on the innate specificity of the sgRNA.34,58,59 In addition, there are potential concerns related to transient 53BP1 inhibition and suppression of the p53 pathway in HSPCs because of tumor suppressor function of p53,60 especially because interference with the p53-mediated DDR that normally removes cells after DNA deleterious events may potentially allow survival of cells with genomic aberrations.61 To ensure a transient effect, both i53 and hGSE were added as mRNA in our study, so they could be easily degraded and their activity could be synchronized with the co-administered Cas9 mRNA activity.
Identifying OT sites by using in vivo genome-wide OT analysis vs in silico computational analysis prediction can yield markedly different results. We compared CHANGE-seq assay with the in silico prediction tools Cas-OFFinder, CCTop, and COSMID, and we found that Cas-OFFinder was the most reliable; it predicted 5 of the 8 top sites (62.5%) identified with CHANGE-seq (supplemental Table 3 ). Sequencing analysis performed on in vitro and in vivo samples showed very low OT activity at the 8 identified top OT sites for the sgRNA used in this study; it also confirmed that our combined approach with i53 and hGSE did not lead to any detectable genomic modifications at the top predicted OT sites compared with untreated samples, which demonstrated the high specificity of this guide RNA. A limitation of our targeted sequencing of predicted sites is the inability to detect large deletions or translocations. However, sophisticated studies using high-throughput genome-wide translocation sequencing or targeted capture deep sequencing previously confirmed that 53BP1 inhibition had no adverse impact on Cas9 specificity.36,62,63 Extensive investigations by Schiroli et al41 also demonstrated a lack of genotoxicities after transient p53 inhibition during CRISPR/Cas9 editing based on the absence of increased chromosomal abnormalities, translocations, or mutational burden associated with the use of GSE56.41,64 Furthermore, the immunodeficient mice in our study did not show any hematologic aberrations or gross tumors at 16 weeks after transplant with gene-edited HSPCs (12 mice for +i53; 24 mice for +i53+hGSE). Molecular studies for additional safety assessment before clinical application may be warranted, and the potential risks need to be weighed against the risks of low correction of engrafting HSPCs in the absence of enhancers.
In summary, we provide the first description of a precise targeted gene insertion approach to functionally correct MAGT1 genetic mutations in XMEN HSPCs and apheresis T lymphocytes. Our data demonstrate that our optimized GE conditions with combined GE enhancers (i53 and hGSE) achieved sustained efficient gene correction in engrafting HSPCs and in T cells. For patients with acute infections, functional gene correction of HSPCs and T cells potentially offers a two-pronged approach for more immediate protection by corrected T cells, whereas latent T-cell differentiation from engrafted gene-edited HSPCs allows for permanent correction. The survival advantage in corrected cells also improves the likelihood of clinical benefit from even low numbers of corrected cells. We observed no detectable OT activity in either in vitro or in vivo engrafting XMEN patient HSPCs after transplant, thus providing important safety data for clinical application of this approach for ex vivo gene therapy or cellular therapy for XMEN patients. These gene-edited enhancers are also widely applicable for improving the levels of engrafting gene-edited HSPCs for HDR-mediated gene correction for potential treatment of many other blood and immune cell diseases.
Acknowledgments
The authors thank their patients, healthy donor volunteers, Laboratory of Clinical Immunology and Microbiology nursing staff, and the staff members of the Dowling Apheresis Unit, Cell Processing Section, and Product Development Section of the National Institutes of Health (NIH), Department of Transfusion Medicine for their contributions to this study.
This study was supported by grants from the NIH (Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (Z01-AI-00988 and 00644; the National Cancer Institute, R00-CA218870; and National Heart, Lung, and Blood Institute, P01-HL142494) (B.P.K.).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Authorship
Contribution: J.B. and S.S.D.R. conceived and performed most of the experiments and wrote the manuscript; T.L., S.K., and C.C. helped with mouse experiments; S.L. and X.W. performed genomic analysis; C.R.L. and S.Q.T. performed and analyzed the CHANGE-seq assay; T.L., E.B., G.V., U.C., C.L.S., and K.D. assisted with functional studies; M.B. and L.D.N. provided advice on the ATO system; B.P.K. helped with sgRNA design; E.B., U.C., and S.S.D.R. designed the rAAV6 donor; R.J.M. designed in vitro transcription templates and mRNA constructs; A.B.C. helped with mRNA manufacturing; J.C.R., M.J.L., G.A.D., and H.L.M. provided expert advice and guidance and helped write the paper; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: B.P.K. is an inventor on patents and has patent applications filed by Partners Healthcare that describe genome engineering technologies, and is an advisor to Acrigen Biosciences and serves as a consultant for Avectas and ElevateBio. R.J.M., A.B.C., and G.A.D. are employees of Cellscript, LLC (Madison, WI). The remaining authors declare no competing financial interests.
Correspondence: Suk See De Ravin, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 10, Rm 5-3816, 10 Center Dr, MSC 1456, Bethesda, MD 20892-1456; e-mail: sderavin@niaid.nih.gov.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal