

Key Points
TFAP4 mutations are found in >10% of cases of Burkitt lymphoma, a hallmark B-cell malignancy driven by high MYC expression.
Mechanistically, c-MYC–induced TFAP4 suppresses tumorigenesis of B cells by limiting self-renewal and promoting differentiation.

Abstract
The proliferative burst of B lymphocytes is essential for antigen receptor repertoire diversification during the development and selective expansion of antigen-specific clones during immune responses. High proliferative activity inevitably promotes oncogenesis, the risk of which is further elevated in B lymphocytes by endogenous gene rearrangement and somatic mutations. However, B-cell–derived cancers are rare, perhaps owing to putative intrinsic tumor-suppressive mechanisms. We show that c-MYC facilitates B-cell proliferation as a protumorigenic driver and unexpectedly coengages counteracting tumor suppression through its downstream factor TFAP4. TFAP4 is mutated in human lymphoid malignancies, particularly in >10% of Burkitt lymphomas, and reduced TFAP4 expression was associated with poor survival of patients with MYC-high B-cell acute lymphoblastic leukemia. In mice, insufficient TFAP4 expression accelerated c-MYC–driven transformation of B cells. Mechanistically, c-MYC suppresses the stemness of developing B cells by inducing TFAP4 and restricting self-renewal of proliferating B cells. Thus, the pursuant transcription factor cascade functions as a tumor suppressor module that safeguards against the transformation of developing B cells.
Introduction
Lymphocytes are one of the most rapidly proliferating cell types in the postnatal organism.1-6 Extensive proliferative burst is essential to form diverse antigen receptor repertoires and facilitate requisite antigen-specific immune responses. In addition to demands for accurate DNA replication during extensive cell division, B lymphocytes are vulnerable to genomic instability as a result of DNA editing by recombination-activating gene (RAG) or activation-induced cytidine deaminase that primarily targets antigen receptor loci but also attacks other sites throughout the genome,7 potentially rendering lymphocytes highly cancer prone. However, cancers derived from developing lymphocytes are rarer than would be expected from their inherent risk, and their incidence is lower than that of common epithelial cancers (eg, 1.7 cases of acute lymphoblastic leukemia [ALL] per 100 000 population annually vs 38-127 cases of colon, lung, and breast cancer per 100 000 population annually ).8 Accordingly, we hypothesized that lymphocytes are protected from transformation by putative tumor-suppressive programs that are tightly linked to their proliferative program. Such programs may preserve the genomic integrity or restrict specific gene-expression programs that would enhance transformation.
The transcription factor c-MYC is a strong oncogenic driver; activating mutations and translocations that deregulate its expression are hallmark features of numerous tumors in humans and mice.9-11 c-MYC is also essential for the proliferation of normal lymphocytes through control of the cell cycle and metabolism.12-15 If lymphocytes activate putative tumor suppressors tied to their proliferation, c-MYC itself may directly induce such tumor suppressor genes as a feedback pathway. Therefore, it is conceivable that perturbation of any such pathways might accelerate the development of lymphoid malignancies induced by aberrant c-MYC expression, although it remains unknown whether such a c-MYC–induced tumor-suppressive mechanism exists.
In this study, we profiled direct c-MYC target genes that are mutated in primary human lymphoid cancers and identified the transcription factor TFAP4 (also known as AP4) as a c-MYC–inducible tumor suppressor in B lymphocytes. Recurrent mutations of TFAP4 are found predominantly in its DNA binding domain in human cancers, including ALL, and a notable proportion of cases of Burkitt lymphoma (BL). In c-MYC–driven mouse B-cell tumor models, TFAP4 functions as a c-MYC–induced haploinsufficient tumor suppressor. Single allele deletion of Tfap4 or specific deletion of a c-MYC binding site in the locus shortened the latency of the MYC transgene-driven tumorigenesis, with frequent loss of heterozygosity (LOH). Mechanistically, c-MYC drives the proliferation of B-cell progenitors, as well as promotes their differentiation, thus restricting the self-renewing capacity of B cells in undifferentiated states through TFAP4-dependent repression of the stemness factor Erg. Finally, reduced expression of TFAP4, despite high c-MYC expression, is associated with poor prognosis of a subset of patients with B-cell ALL (B-ALL). These results collectively show that the c-MYC–TFAP4 axis has dual roles in supporting the clonal expansion of normal lymphocytes16,17 and paradoxically, suppresses the transformation of proliferating B cells.
Methods
Mice
Tfap4−/−,18Tfap4F/F,16ErgF/F,19Cd79a-icre,20 and Myc-GFP21 mice were described previously and were backcrossed to the C57BL/6N background for >10 generations. Eμ-Myc,22Ighg1-cre,23Rosa26CAG-MYC,24 and Rosa26Pik3ca*25 mice were from The Jackson Laboratory. C57BL/6N and B6-Ly5.1 mice were from Charles River. All mouse experiments were conducted according to the protocol approved by the Washington University Animal Studies Committee.
Tumorigenesis studies
Eμ-Myc mice were monitored weekly for palpable tumor development and body weight loss. Mice were euthanized when tumor sizes exceeded 20 mm in diameter or mice exhibited >20% body weight loss or paralysis. For transplantation of retrovirally transduced B cells, CD19+ bone marrow (BM) cells, isolated using anti-CD19 MicroBeads (Miltenyi Biotec) from 6- to 10-week-old mice, were infected with viral supernatants, followed by IV transfer of 1 × 106 cells into sublethally irradiated (5.5 Gy) mice. For in vivo persistence analysis, transduced cells were transferred into congenic mice at an ∼1:1 ratio of Tfap4+/−, Tfap4+/−Erg+/−, or Erg+/− cells to wild-type (WT) cells.
Human BL and TARGET data analysis
Gene expression data from BL samples26 were downloaded from the European Genome-Phenome Archive (EGAS00001003778). Data were analyzed using Galaxy as follows: counts per gene for each sample were obtained by running featureCounts on GRCh38-aligned BAM files and normalized using DESeq2. The normalized data were imported into the Phantasus application27 for further analysis. After log2 transformation and quantile normalization, ENTREZ-annotated genes were filtered for mean normalized counts >2 across samples. Data for B-ALL samples were downloaded from TARGET phase II, and gene expression was analyzed in Phantasus. Differentially expressed genes (DEGs) in both patient cohorts were analyzed using a limma tool, followed by gene set enrichment analysis (GSEA).
Statistical analysis
Survival data were compared using the log-rank test with adjustment for multiple comparisons when necessary. P values were calculated using an unpaired 2-tailed Student t test for 2-group comparisons and 1-way analysis of variance (ANOVA) for multigroup comparisons with the Tukey post hoc test or Kruskal-Wallis test with Dunn’s multiple comparison test. Human microarray data from previously published studies were compared using Mann-Whitney’s U test for 2-group comparisons and the Kruskal-Wallis test for multigroup comparisons. All statistical analyses were done using Prism. Adjusted values of P < .05 were considered significant.
Results
The c-MYC downstream transcription factor TFAP4 is mutated in primary human cancers
To determine whether c-MYC engages tumor-suppressive programs in B lymphocytes, we initially defined c-MYC–dependent genes in developing B cells between Eμ-Myc and littermate WT B220+ immunoglobulin M (IgM)− pro/pre-B cells. We combined this analysis with gene expression profiles of mouse germinal center B (GCB) cells13 and identified 107 genes that were commonly dependent on c-MYC in pro/pre-B and GCB cells (Figure 1A-C). The candidate genes were prioritized based on known roles in gene regulation, direct c-MYC targets,16,17,28 and mutation frequencies in human hematopoietic malignancies using the COSMIC (Catalogue of Somatic Mutations In Cancer) database. Tfap4, encoding a bHLH transcription factor, was identified as a gene meeting all of the criteria (Figure 1D). We confirmed that TFAP4 expression was increased in B220+ IgM− BM cells from Eμ-Myc mice compared with those from littermate WT mice (Figure 1E).
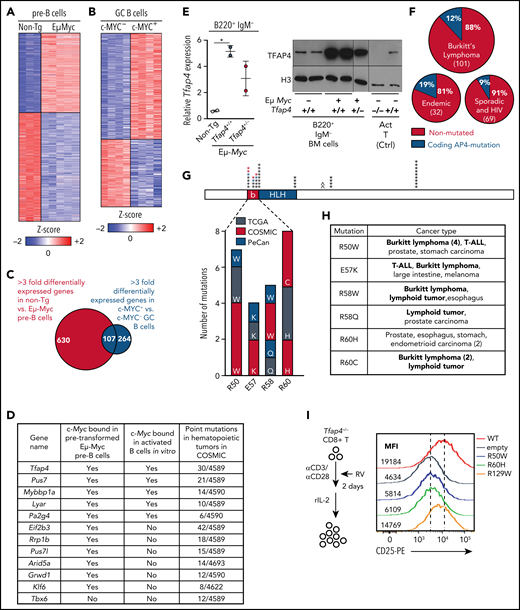
The transcription factor TFAP4 is induced by MYC in developing B cells and GCB cells and is mutated in lymphoid cancers. (A) Z score heat map showing expression of genes that are differentially expressed at least threefold by pro/pre-B cells from Eμ-Myc−Tfap4+/+ (non-Tg; n = 3) and Eμ-Myc Tfap4+/+ (n = 5) mice, as determined by RNA sequencing. (B) Z score heat map showing expression of genes that are differentially expressed at least threefold by c-MYC+ vs c-MYC− mouse splenic GCB cells, as determined by microarray (GSE38304). (C) Overlap of DEGs in (A) and (B). (D) Overlapping genes from panel C involved in gene regulation with direct c-MYC binding to each genomic region and mutation frequencies in hematopoietic tumors in COSMIC. The list is sorted by the numbers of point mutations in hematopoietic tumors after prioritization of c-MYC–bound genes. (E) Quantitative reverse transcription polymerase chain reaction (left panel) and immunoblot analysis (right panel) of Tfap4 mRNA and TFAP4 protein in BM B220+ IgM− cells of Eμ-Myc−Tfap4+/+ (non-Tg) and Eμ-Myc+ Tfap4+/+ and Tfap4+/− mice. Tfap4 mRNA expression was normalized to spike-in control RNA, ERCC-00108. Histone H3 serves as a loading control in immunoblot. (F) Frequency of coding TFAP4 mutations registered across BL subtypes.26 (G) Mapping of recurrent (≥4 independent) somatic missense mutations of TFAP4 identified in primary human tumors from TCGA, PeCan, and COSMIC databases. Each mutation is shown by an asterisk and is mapped to its position in the TFAP4 protein; PeCan lymphoid tumors are shown in blue, and Duke BL cases are shown in red. The basic region (b) functioning as a DNA binding domain and the helix-loop-helix (HLH) domain are shown. A caret (^) indicates that the R129W mutation is found outside of this region. (H) List of recurrent somatic mutations in the DNA binding region and their cancer types in panel G with hematopoietic malignancies shown in bold. (I) Assay to determine the function of somatic TFAP4 variants in upregulation of CD25, which is a direct TFAP4 target (supplemental Figure 1C-E), in Tfap4−/− CD8 T cells (left panel). Expression of CD25 in Tfap4−/− CD8 T cells retrovirally expressing each TFAP4 somatic variant (right panel). Data are representative of 3 independent experiments . *P < .05 by 1-way ANOVA. Ctrl, control; MFI, median fluorescence intensity; rIL-2, recombinant interleukin-2; RV; retrovirus; α, anti-.
The transcription factor TFAP4 is induced by MYC in developing B cells and GCB cells and is mutated in lymphoid cancers. (A) Z score heat map showing expression of genes that are differentially expressed at least threefold by pro/pre-B cells from Eμ-Myc−Tfap4+/+ (non-Tg; n = 3) and Eμ-Myc Tfap4+/+ (n = 5) mice, as determined by RNA sequencing. (B) Z score heat map showing expression of genes that are differentially expressed at least threefold by c-MYC+ vs c-MYC− mouse splenic GCB cells, as determined by microarray (GSE38304). (C) Overlap of DEGs in (A) and (B). (D) Overlapping genes from panel C involved in gene regulation with direct c-MYC binding to each genomic region and mutation frequencies in hematopoietic tumors in COSMIC. The list is sorted by the numbers of point mutations in hematopoietic tumors after prioritization of c-MYC–bound genes. (E) Quantitative reverse transcription polymerase chain reaction (left panel) and immunoblot analysis (right panel) of Tfap4 mRNA and TFAP4 protein in BM B220+ IgM− cells of Eμ-Myc−Tfap4+/+ (non-Tg) and Eμ-Myc+ Tfap4+/+ and Tfap4+/− mice. Tfap4 mRNA expression was normalized to spike-in control RNA, ERCC-00108. Histone H3 serves as a loading control in immunoblot. (F) Frequency of coding TFAP4 mutations registered across BL subtypes.26 (G) Mapping of recurrent (≥4 independent) somatic missense mutations of TFAP4 identified in primary human tumors from TCGA, PeCan, and COSMIC databases. Each mutation is shown by an asterisk and is mapped to its position in the TFAP4 protein; PeCan lymphoid tumors are shown in blue, and Duke BL cases are shown in red. The basic region (b) functioning as a DNA binding domain and the helix-loop-helix (HLH) domain are shown. A caret (^) indicates that the R129W mutation is found outside of this region. (H) List of recurrent somatic mutations in the DNA binding region and their cancer types in panel G with hematopoietic malignancies shown in bold. (I) Assay to determine the function of somatic TFAP4 variants in upregulation of CD25, which is a direct TFAP4 target (supplemental Figure 1C-E), in Tfap4−/− CD8 T cells (left panel). Expression of CD25 in Tfap4−/− CD8 T cells retrovirally expressing each TFAP4 somatic variant (right panel). Data are representative of 3 independent experiments . *P < .05 by 1-way ANOVA. Ctrl, control; MFI, median fluorescence intensity; rIL-2, recombinant interleukin-2; RV; retrovirus; α, anti-.
Somatic mutations of TFAP4 were found in lymphoid and other cancers registered in COSMIC, TCGA (The Cancer Genome Atlas), and PeCan (Pediatric Cancer Genomic Data Portal). Notably, 5 of 6 PeCan cases with TFAP4 mutations were found in lymphoid tumors (3 BL, 1 B-ALL, and 1 T-cell ALL [T-ALL]). Although TFAP4 mutations in these databases were rare (288/37 420 unique samples in COSMIC, 65/10 202 in TCGA, and 6/2578 in PeCan), analyses identified missense TFAP4 mutations in 12 of 101 cases in the BL data set:26 6 of 32 endemic BLs and 6 of 69 sporadic and HIV+ cases (Figure 1F; supplemental Figure 1A-B, available on the Blood Web site). Across these data sets, the majority of recurrent somatic mutations (≥4 independent cases), including 4 of 6 cases in PeCan lymphoid tumors and 3 additional BL cases, were mapped to the DNA binding domain (basic region)29 (Figure 1G-H). A single amino acid substitution in the DNA binding residue was sufficient to compromise its transactivating potential, because expression of CD25/Il2ra, a direct TFAP4 target gene, was reduced in Tfap4−/− CD8 T cells retrovirally complemented with the R50W or R60H variant compared with WT-TFAP4 or the R129W variant harboring a mutation outside of the basic region (Figure 1I; supplemental Figure 1C-F). These results suggest that the somatic mutations found in the basic region impair TFAP4 function and that the dysfunction of TFAP4 contributes to oncogenesis in lymphoid malignancies.
TFAP4 is a cell-intrinsic tumor suppressor
Because Tfap4 is a c-MYC–inducible gene16,17,30 and many tumors associated with TFAP4 mutations express high levels of c-MYC, we next determined whether TFAP4 modulates c-MYC–induced tumorigenesis in B cells. We first used the Eμ-Myc mouse model, in which Myc is expressed under the control of the Igh μ enhancer, mimicking the t(8;14) IGH-MYC translocation.22 In this model, c-MYC is overexpressed in developing B cells starting at the pro-B–cell stage, leading to B-cell malignancy and nearly all mice dying within a year (Figure 2A). In striking contrast, haploinsufficiency of Tfap4 in Eμ-Myc mice accelerated tumorigenesis, with all mice dying by day 92, whereas homozygous Tfap4 deletion further accelerated tumorigenesis (Figure 2A). In 40% of tumors in Eμ-Myc mice, B220+ cells exhibited an immature phenotype, expressing Rag1 and VpreB1 (Figure 2B; supplemental Figure 2A-B), consistent with previous studies.31,32 In contrast, 93% of Eμ-Myc Tfap4+/− mice formed tumors with the immature phenotype (Figure 2B). The skewing of tumors toward the immature phenotype suggests that pro/pre-B cells with elevated c-MYC, but reduced TFAP4, become transformed with shorter latency after c-MYC is turned on in pro-B cells. The numbers of developing B cells, their short-term proliferation, and annexin V binding at the population level were comparable between Tfap4-haploinsufficient and Tfap4-proficient Eμ-Myc mice before tumor formation (supplemental Figure 2C-F). No survival difference was observed between Tfap4+/+ and Tfap4+/− Eμ-Myc pro/pre-B cells ex vivo (supplemental Figure 2G-H).
![TFAP4 is a c-MYC–induced cell-intrinsic tumor suppressor in developing B cells. (A) Survival analysis of Eμ-Myc mice crossed to Tfap4+/+ mice (n = 68), Tfap4+/− mice (n = 50), or Tfap4−/− mice (n = 6). The median survival was 134, 62, and 36 days, respectively. (B) Frequencies of surface Ig−Rag1+ VpreB1+ (immature) and Ig+Rag1− VpreB1− (mature) B-cell tumors in Eμ-Myc mice with distinct Tfap4 genotypes (Eμ-Myc Tfap4+/+, n = 10; Eμ-Myc Tfap4+/−, n = 14; Eμ-Myc Tfap4−/−, n = 2). (C) Survival of Eμ-Myc mice with Cd79aicre–mediated deletion of the Tfap4 allele in developing B cells (Tfap4F/+ or Tfap4F/F cre− [n = 59], median survival, 104 days; Tfap4+/+ Cd79aicre/+ [n = 16], median survival, 113 days; Tfap4F/+ Cd79aicre/+ [n = 17], median survival, 59 days; Tfap4F/F Cd79aicre/+ [n = 6], median survival, 42 days). (D) Tumorigenesis of Tfap4+/+ or Tfap4−/− BM CD19+ cells retrovirally transduced with mouse c-MYC (T58A) and adoptively transferred into sublethally irradiated CD45.1 mice (Tfap4+/+ [n = 10], median survival, undefined days; Tfap4−/− [n = 12], median survival, 83.5 days;). Two independent experiments were combined. (E) Survival of mice receiving Tfap4+/+ or Tfap4−/− BM CD19+ cells transduced with human P190 bcr-abl–expressing retrovirus and adoptively transferred into sublethally irradiated CD45.1 mice (Tfap4+/+ [n = 9], median survival, 32 days; Tfap4−/− [n = 10], median survival, 19.5 days). Two independent experiments were combined. (F) Survival of lethally irradiated CD45.1 mice reconstituted with BM cells from Cγ1-cre R26StopLSL-MYC × R26StopLSL-P110* mice of the indicated Tfap4 genotypes following immunization with sheep red blood cells. Data are combined from 4 independent experiments. Tfap4+/+ (n = 12), median survival, 88 days; Tfap4F/+ (n = 14), median survival, 74.5 days; Tfap4F/F (n = 15), median survival: 27 days. **P < .01, ***P < .001, log-rank test adjusted for multiple comparisons. RV, retrovirus.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/24/10.1182_blood.2021011711/5/m_bloodbld2021011711f2.png?Expires=1769079987&Signature=j9DkF7WLdMN-TxTTl5urqglgn-skiEnnka8BLow-q25~NGQR8xshwIc-8pbpEHnXH~fDuXaM4iaSdfZQ5Vfn7G274VtDXEukvrMizey~qN-X7VcrsMYRssgj8XMyyo~DCNmDddb~yyt1chkj4hr8UT8ge6xG85kzbO4lg1tk9t3kErxGU-7ObqmsD~4ElWYqimSIE0QWVOdiVQqto8CjowZZaGjZBc9VZZcfp5veP7J4xW6C28D7l-OqjNQ6SQ~JRM9LqO2zv6p0HmLwL4mY4qm4BYlxwVUZVvbrYXMmWFgH4cn4iFTl3YGEhFeVbIB9Y~z27rGtkSrMPdS1ATqwPQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TFAP4 is a c-MYC–induced cell-intrinsic tumor suppressor in developing B cells. (A) Survival analysis of Eμ-Myc mice crossed to Tfap4+/+ mice (n = 68), Tfap4+/− mice (n = 50), or Tfap4−/− mice (n = 6). The median survival was 134, 62, and 36 days, respectively. (B) Frequencies of surface Ig−Rag1+ VpreB1+ (immature) and Ig+Rag1− VpreB1− (mature) B-cell tumors in Eμ-Myc mice with distinct Tfap4 genotypes (Eμ-Myc Tfap4+/+, n = 10; Eμ-Myc Tfap4+/−, n = 14; Eμ-Myc Tfap4−/−, n = 2). (C) Survival of Eμ-Myc mice with Cd79aicre–mediated deletion of the Tfap4 allele in developing B cells (Tfap4F/+ or Tfap4F/F cre− [n = 59], median survival, 104 days; Tfap4+/+ Cd79aicre/+ [n = 16], median survival, 113 days; Tfap4F/+ Cd79aicre/+ [n = 17], median survival, 59 days; Tfap4F/F Cd79aicre/+ [n = 6], median survival, 42 days). (D) Tumorigenesis of Tfap4+/+ or Tfap4−/− BM CD19+ cells retrovirally transduced with mouse c-MYC (T58A) and adoptively transferred into sublethally irradiated CD45.1 mice (Tfap4+/+ [n = 10], median survival, undefined days; Tfap4−/− [n = 12], median survival, 83.5 days;). Two independent experiments were combined. (E) Survival of mice receiving Tfap4+/+ or Tfap4−/− BM CD19+ cells transduced with human P190 bcr-abl–expressing retrovirus and adoptively transferred into sublethally irradiated CD45.1 mice (Tfap4+/+ [n = 9], median survival, 32 days; Tfap4−/− [n = 10], median survival, 19.5 days). Two independent experiments were combined. (F) Survival of lethally irradiated CD45.1 mice reconstituted with BM cells from Cγ1-cre R26StopLSL-MYC × R26StopLSL-P110* mice of the indicated Tfap4 genotypes following immunization with sheep red blood cells. Data are combined from 4 independent experiments. Tfap4+/+ (n = 12), median survival, 88 days; Tfap4F/+ (n = 14), median survival, 74.5 days; Tfap4F/F (n = 15), median survival: 27 days. **P < .01, ***P < .001, log-rank test adjusted for multiple comparisons. RV, retrovirus.
TFAP4 is a c-MYC–induced cell-intrinsic tumor suppressor in developing B cells. (A) Survival analysis of Eμ-Myc mice crossed to Tfap4+/+ mice (n = 68), Tfap4+/− mice (n = 50), or Tfap4−/− mice (n = 6). The median survival was 134, 62, and 36 days, respectively. (B) Frequencies of surface Ig−Rag1+ VpreB1+ (immature) and Ig+Rag1− VpreB1− (mature) B-cell tumors in Eμ-Myc mice with distinct Tfap4 genotypes (Eμ-Myc Tfap4+/+, n = 10; Eμ-Myc Tfap4+/−, n = 14; Eμ-Myc Tfap4−/−, n = 2). (C) Survival of Eμ-Myc mice with Cd79aicre–mediated deletion of the Tfap4 allele in developing B cells (Tfap4F/+ or Tfap4F/F cre− [n = 59], median survival, 104 days; Tfap4+/+ Cd79aicre/+ [n = 16], median survival, 113 days; Tfap4F/+ Cd79aicre/+ [n = 17], median survival, 59 days; Tfap4F/F Cd79aicre/+ [n = 6], median survival, 42 days). (D) Tumorigenesis of Tfap4+/+ or Tfap4−/− BM CD19+ cells retrovirally transduced with mouse c-MYC (T58A) and adoptively transferred into sublethally irradiated CD45.1 mice (Tfap4+/+ [n = 10], median survival, undefined days; Tfap4−/− [n = 12], median survival, 83.5 days;). Two independent experiments were combined. (E) Survival of mice receiving Tfap4+/+ or Tfap4−/− BM CD19+ cells transduced with human P190 bcr-abl–expressing retrovirus and adoptively transferred into sublethally irradiated CD45.1 mice (Tfap4+/+ [n = 9], median survival, 32 days; Tfap4−/− [n = 10], median survival, 19.5 days). Two independent experiments were combined. (F) Survival of lethally irradiated CD45.1 mice reconstituted with BM cells from Cγ1-cre R26StopLSL-MYC × R26StopLSL-P110* mice of the indicated Tfap4 genotypes following immunization with sheep red blood cells. Data are combined from 4 independent experiments. Tfap4+/+ (n = 12), median survival, 88 days; Tfap4F/+ (n = 14), median survival, 74.5 days; Tfap4F/F (n = 15), median survival: 27 days. **P < .01, ***P < .001, log-rank test adjusted for multiple comparisons. RV, retrovirus.
To determine whether TFAP4-mediated supression of Eμ-Myc tumors is cell-intrinsic, we deleted 1 or 2 Tfap4F alleles in pro-B cells using Cd79aicre, recapitulating the accelerated tumorigenesis and immunophenotype observed in germline Tfap4-deficient mice (Figure 2C; supplemental Figure 2I). We also tested whether TFAP4 functions as a suppressor of c-MYC–induced B cell malignancies in additional mouse models, because almost all B cells express aberrant MYC throughout their development in Eμ-Myc mice. First, we retrovirally expressed c-MYC in CD19+ BM cells from Tfap4−/− or Tfap4+/+ mice, followed by transfer to sublethally irradiated mice. In this model, we typically achieve transduction of ∼10% of pro/pre-B cells (data not shown), and tumor formation by c-MYC–transduced pre-B cells is dependent on a defect in a tumor suppressor.33,34 In contrast to control mice receiving c-MYC–transduced Tfap4+/+ CD19+ cells, the majority of mice receiving c-MYC–transduced Tfap4−/− CD19+ BM cells died from pre-B–cell tumors (Figure 2D; supplemental Figure 2J). We also observed accelerated B-ALL disease progression induced by ectopic expression of p190 bcr-abl, which indirectly upregulates Myc, in CD19+ BM cells from Tfap4−/− mice compared with those from Tfap4+/+ mice (Figure 2E; supplemental Figure 2K). In addition to these immature B-cell malignancies, we observed accelerated death in the absence of TFAP4 in a mature B-cell lymphoma model induced by c-MYC and a constitutively active phosphatidylinositol 3-kinase24 (Figure 2F). These data establish a cell-intrinsic requirement for TFAP4 in the suppression of MYC-mediated B-cell cancers in multiple mouse models.
c-MYC bound 2 consensus sites within the H3K27ac-marked intronic region at the Tfap4 locus in Eμ-Myc B220+ BM cells (supplemental Figure 3A). To determine whether direct activation of Tfap4 by c-MYC binding to this enhancer is necessary for tumor suppression, we deleted a 1-kb intronic region containing the 2 consensus sites using CRISPR/Cas9 (supplemental Figure 3A). Heterozygous deletion of the 1-kb region in Eμ-Myc mice recapitulated accelerated tumorigenesis (supplemental Figure 3B-C). This might be caused by the loss of other transcription factors binding at the enhancer or indirectly by altered expression of the neighboring genes. However, a more specific single-allele deletion of 1 MYC binding site also shortened survival of these mice compared with Eμ-Myc mice harboring WT Tfap4 loci (supplemental Figure 3B), whereas expression of 2 genes around the Tfap4 locus, or Runx1 located on the same chromosome, was unaffected (supplemental Figure 3C). Thus, direct activation of Tfap4 by c-MYC is essential for tumor suppression.
LOH of TFAP4 confers interclonal competitiveness for tumor outgrowth
Tfap4 messenger RNA (mRNA) expression was comparable between pro/pre-B cells before transformation and pro/pre-B tumor cells in Eμ-Myc Tfap4+/+ mice (Figure 3A). However, approximately one third of pro/pre-B tumors from Eμ-Myc Tfap4+/− mice expressed substantially lower Tfap4 mRNA compared with pretransformed pro/pre-B cells (Figure 3A, dashed rectangle). This reduced Tfap4 expression was caused by LOH in these tumors (Figure 3B). A genomic region encompassing exons 2 through 4, deleted in the Tfap4– allele, was detectable in all tumors from Eμ-Myc Tfap4+/+ mice and a subset of tumors from Eμ-Myc Tfap4+/− mice. By contrast, this genomic region was lost in tumor cells with low Tfap4 expression from Eμ-Myc Tfap4+/− mice (Figure 3B, dashed rectangle). LOH of Tfap4 was also confirmed by loss of mCherry (mC) fluorescence in Eμ-Myc tumors harboring 1 null and 1 functional TFAP4-mC fusion knock-in allele17 (Tfap4mC/−) (Figure 3C-D). These results indicate that, although TFAP4 downregulation is unnecessary for transformation, rare clones undergoing LOH at the Tfap4 locus predominantly contribute to tumorigenesis.
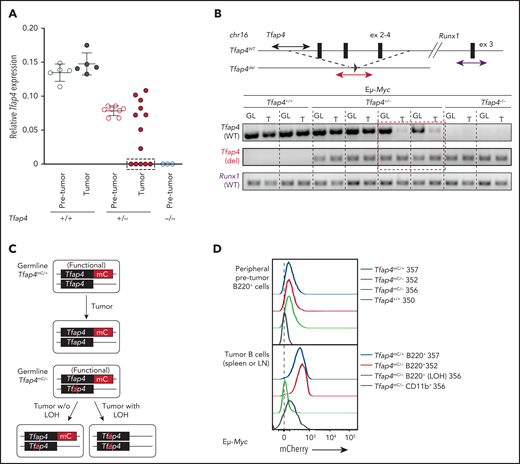
Low levels of TFAP4 endow interclonal competition of pretransformed B-cell precursors with aberrant c-MYC expression. (A) Expression of Tfap4 mRNA in pretransformed B220+IgM− BM (Pre-tumor) or B220+ IgM− tumor (Tumor) cells harvested from Eμ-Myc mice with the indicated Tfap4 genotypes. Tfap4 mRNA expression, measured by quantitative reverse transcription polymerase chain reaction (PCR), was normalized to Hprt1 expression. (B) Genomic PCR detecting the region of the Tfap4 allele encompassing exons 2-4 in tumor DNA (T) and tail DNA (GL, germline) of each corresponding mouse. A genomic region containing Runx1 located on the same chromosome was used as an internal control. Three of 5 lymphomas with low Tfap4 mRNA expression were analyzed by genomic PCR; the red dashed box indicates 2 representative samples with LOH of Tfap4 in tumors. (C) Diagram showing the detection of LOH using Tfap4-mC reporter knock-in mice. (D) Detection of TFAP4-mC fusion protein in peripheral blood B cells from healthy 4-week old Eμ-Myc Tfap4mC/+ and Eμ-Myc Tfap4mC/– mice (upper panel). Loss of TFAP4-mC expression in Eμ-Myc Tfap4mC/– tumor B cells with LOH or CD11b+ cells as internal mC– control (lower panel). The numbers to the right represent unique mouse IDs. chr16, chromosome 16; ex, exon; w/o, without.
Low levels of TFAP4 endow interclonal competition of pretransformed B-cell precursors with aberrant c-MYC expression. (A) Expression of Tfap4 mRNA in pretransformed B220+IgM− BM (Pre-tumor) or B220+ IgM− tumor (Tumor) cells harvested from Eμ-Myc mice with the indicated Tfap4 genotypes. Tfap4 mRNA expression, measured by quantitative reverse transcription polymerase chain reaction (PCR), was normalized to Hprt1 expression. (B) Genomic PCR detecting the region of the Tfap4 allele encompassing exons 2-4 in tumor DNA (T) and tail DNA (GL, germline) of each corresponding mouse. A genomic region containing Runx1 located on the same chromosome was used as an internal control. Three of 5 lymphomas with low Tfap4 mRNA expression were analyzed by genomic PCR; the red dashed box indicates 2 representative samples with LOH of Tfap4 in tumors. (C) Diagram showing the detection of LOH using Tfap4-mC reporter knock-in mice. (D) Detection of TFAP4-mC fusion protein in peripheral blood B cells from healthy 4-week old Eμ-Myc Tfap4mC/+ and Eμ-Myc Tfap4mC/– mice (upper panel). Loss of TFAP4-mC expression in Eμ-Myc Tfap4mC/– tumor B cells with LOH or CD11b+ cells as internal mC– control (lower panel). The numbers to the right represent unique mouse IDs. chr16, chromosome 16; ex, exon; w/o, without.
A c-MYC target gene signature is commonly enriched in Eμ-Myc Tfap4+/− tumors and TFAP4-mutated BL in humans
To gain insights into the molecular mechanisms of c-MYC–TFAP4–dependent tumor suppression, we profiled the gene expression of pretransformed B220+ IgM− pro/pre-B cells, validated by unbiased expression of Ighv transcripts (supplemental Figure 4A), from Eμ-Myc Tfap4+/− mice and control Eμ-Myc Tfap4+/+ mice at 3 to 4 weeks of age. Because of rapid tumor development, analysis of pretransformed Eμ-Myc Tfap4−/− pro/pre-B cells was excluded. Gross transcriptomic differences determined by principal component analysis and the expression of genes related to apoptosis, proliferation, and senescence were not detected between Eμ-Myc Tfap4+/− cells and Eμ-Myc Tfap4+/+ cells (supplemental Figure 4B-D). At the gene level, 29 genes were differentially expressed by ≥1.8-fold. Among these, 20 genes were directly bound by TFAP4, as confirmed by chromatin immunoprecipitation sequencing (current study), and 9 of the 20 genes were shared TFAP4 and MYC targets28 in Eμ-Myc B cells (Figure 4A-C). Despite the lack of clear genotype-associated clustering (supplemental Figure 4E), an expression pattern of 17 of these 29 genes was preserved in Tfap4-deficient tumors, suggesting that their expression is maintained, or selected for, during tumorigenesis (supplemental Figure 4F).
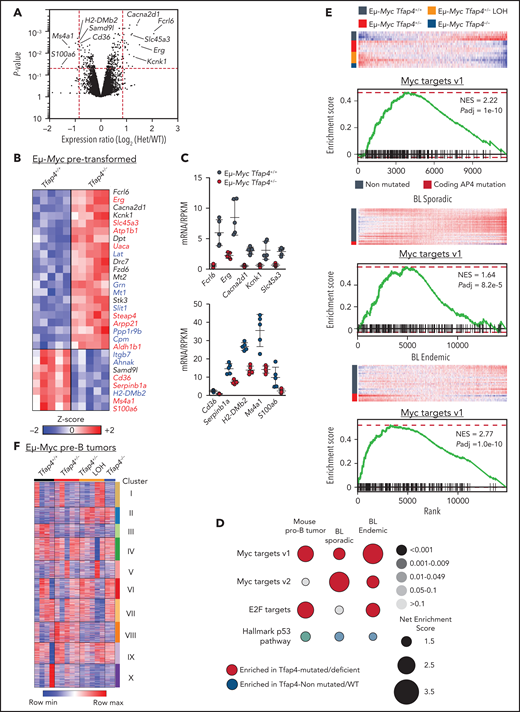
Gene expression changes associated with c-MYC target gene signature are enriched in mouse Eμ-Myc Tfap4+/− tumors and TFAP4-mutated BL patient samples. (A) Volcano plot showing gene expression changes between pretransformed Eμ-Myc Tfap4+/+ and Eμ-Myc Tfap4+/− B220+ IgM− BM cells. Log2-fold change and P values were calculated using a limma R package through the Phantasus application. Red dashed lines indicate cutoff thresholds for fold-change >1.8 and P < .05. The top 5 most DEGs in either direction are labeled. (B) Annotation and z score heat map of genes differentially expressed by >1.8-fold in (A). Genes bound by TFAP4 or cobound by TFAP4 and MYC, as determined by chromatin immunoprecipitation sequencing, are shown in red and blue, respectively. (C) Expression of DEGs, defined in (B), by pretransformed pro/pre-B cells from mice with the indicated Tfap4 genotypes. Upper panel: upregulated genes; lower panel: downregulated genes. (D) GSEA of Tfap4-deficient/mutated vs Tfap4-WT/nonmutated mouse tumor B cells or human BL samples relative to the indicated gene set signatures. Dot sizes, colors, and color gradients indicate the net enrichment score, positive or negative enrichment, and adjusted P value, respectively. (E) GSEA illustrating the selective upregulation of MYC target genes in Tfap4-deficient/haploinsufficient mouse tumors (top panels) and TFAP4-mutated human sporadic (middle panels) or endemic (bottom panels) BL samples. (F) K-means clustering (10 clusters) of genes expressed in pro/pre-B tumor cells from Eμ-Myc mice with indicated Tfap4 genotypes. Het, heterozygous; max, maximum; min, minimum; NES, net enrichment score; Padj, adjusted P value; RPKM, reads per kilobase of transcript, per million mapped reads.
Gene expression changes associated with c-MYC target gene signature are enriched in mouse Eμ-Myc Tfap4+/− tumors and TFAP4-mutated BL patient samples. (A) Volcano plot showing gene expression changes between pretransformed Eμ-Myc Tfap4+/+ and Eμ-Myc Tfap4+/− B220+ IgM− BM cells. Log2-fold change and P values were calculated using a limma R package through the Phantasus application. Red dashed lines indicate cutoff thresholds for fold-change >1.8 and P < .05. The top 5 most DEGs in either direction are labeled. (B) Annotation and z score heat map of genes differentially expressed by >1.8-fold in (A). Genes bound by TFAP4 or cobound by TFAP4 and MYC, as determined by chromatin immunoprecipitation sequencing, are shown in red and blue, respectively. (C) Expression of DEGs, defined in (B), by pretransformed pro/pre-B cells from mice with the indicated Tfap4 genotypes. Upper panel: upregulated genes; lower panel: downregulated genes. (D) GSEA of Tfap4-deficient/mutated vs Tfap4-WT/nonmutated mouse tumor B cells or human BL samples relative to the indicated gene set signatures. Dot sizes, colors, and color gradients indicate the net enrichment score, positive or negative enrichment, and adjusted P value, respectively. (E) GSEA illustrating the selective upregulation of MYC target genes in Tfap4-deficient/haploinsufficient mouse tumors (top panels) and TFAP4-mutated human sporadic (middle panels) or endemic (bottom panels) BL samples. (F) K-means clustering (10 clusters) of genes expressed in pro/pre-B tumor cells from Eμ-Myc mice with indicated Tfap4 genotypes. Het, heterozygous; max, maximum; min, minimum; NES, net enrichment score; Padj, adjusted P value; RPKM, reads per kilobase of transcript, per million mapped reads.
To further examine gene expression program modules that were altered specifically in Tfap4-deficient pre-B tumors compared with Tfap4-proficient pre-B tumors, we conducted GSEA. Among curated gene sets in the Molecular Signature Database (https://www.gsea-msigdb.org/gsea/msigdb/),35-37 genes related to MYC targets and E2F targets were significantly enriched in Tfap4-deficient/haploinsufficient tumors (Figure 4D-E). Gene sets associated with MYC and E2F targets were also significantly enriched in human BL samples harboring somatic TFAP4 mutations (Figure 4D-E). These findings suggest that TFAP4 functions as a negative feedback factor that restricts proliferative states of tumorigenic cells associated with high c-MYC and E2F activity. Unsupervised clustering of DEGs in pre-B–cell tumors with distinct Tfap4 genotypes revealed 2 clusters specifically associated with reduced Tfap4 expression (Figure 4F, clusters II/III). These clusters were enriched for genes associated with cancer/p53-related pathways (supplemental Figure 4H). In addition, a gene signature associated with an upregulated KRAS signaling pathway, which has been associated with the Eμ-Myc model,38-40 was enriched specifically in Eμ-Myc Tfap4+/− tumors (Figure 4F, cluster VIII; supplemental Figure 4H). Consistently, whole-exome sequencing and targeted sequencing of the tumor samples, which were mostly clonal or oligoclonal (supplemental Figure 5A), revealed frequent Kras mutations in Eμ-Myc Tfap4+/− tumors, despite similar overall mutational burdens (supplemental Figure 5B-D). Overall, transcriptomic and genetic data suggest that TFAP4-dependent tumor suppression is mediated by the regulation of persistent engagement of cell proliferation via specific gene pathways rather than protection against global genomic instability.
Restriction of the proto-oncogene Erg is required for tumor suppression by TFAP4
TFAP4 supports the continued proliferation of normal lymphocytes by supplementing an early decay of c-MYC by maintaining the expression of numerous c-MYC target genes, but it is dispensable when c-MYC is expressed.16,17 However, during tumorigenesis, our data showed that TFAP4 counteracts c-MYC–mediated transformation of developing B cells, likely through control of its unique targets. Among the unique TFAP4 targets in Figure 4B, Erg, encoding an ETS-family transcription factor, has been implicated in hematopoietic malignancies. Erg is also necessary to maintain the stemness of hematopoietic stem cells (HSCs).41 ERG overexpression in mouse hematopoietic progenitor cells causes lymphoid and megakaryocytic cancers.42-45 In humans, although impaired ERG function has been associated with a small subset of B-ALL specifically harboring a DUX4 rearrangement,45,46ERG was overexpressed in a majority of B-ALL cases compared with other tumor types and normal BM cells across multiple studies47-51 (supplemental Figure 6A-E). Despite the limited sample number with gene expression data available, a human T-ALL case with a TFAP4E57K substitution and an LMO2-LYL1 translocation in PeCan expressed ERG at a level higher than the 95% confidence interval of ERG expression in other T-ALL samples with an LMO2-LYL1 or LMO1-LMO2 translocation (supplemental Figure 6F-G), suggesting that elevated ERG is associated with functionally defective TFAP4.
In Eμ-Myc Tfap4+/− pretransformed pro/pre-B cells, Erg expression was elevated by 3.8-fold compared with controls (Figure 5A). Erg was further upregulated in pro/pre-B tumors lacking TFAP4 (Figure 5B). TFAP4 bound an intronic region of the Erg locus, colocalizing with H3K27ac-marked accessible chromatin in Eμ-Myc B cells, whereas c-MYC binding was not detected in the genomic region encompassing the Erg locus (Figure 5C). These results suggest that Erg is a direct TFAP4 target. Erg is coexpressed with Myc and Tfap4 in pro-B cells before the Igh checkpoint at the population level but is downregulated in proliferating large pre-B cells as they differentiate into small pre-B cells (Figure 5D). However, within c-kit+ pro-B cells, expression of c-MYC protein correlates inversely with that of Erg mRNA, with Erg expression enriched in c-MYC− pro-B cells and significantly downregulated as cells upregulate c-MYC during the transition to large pre-B cells (Figure 5E). Therefore, we hypothesized that Erg must be regulated in c-MYC+ B cells in a TFAP4-dependent manner to restrict the risk of tumorigenesis, because c-MYC and ERG are required for B-cell development but are also oncogenic. Accordingly, Erg was elevated in Tfap4−/− vs Tfap4+/+ c-MYC+ pro-B cells (Figure 5F). Furthermore, TFAP4-mediated Erg suppression was even more apparent in Eμ-Myc mice, because Erg was completely suppressed in Eμ-Myc vs WT B220+ IgM− cells (Figure 5G). Erg suppression was partially reversed and completely lost in Eμ-Myc Tfap4+/− and Eμ-Myc Tfap4−/− pretransformed pro/pre-B cells, respectively (Figure 5G). These results indicate that TFAP4 prevents simultaneous expression of the 2 proto-oncogenes, c-MYC and ERG, in differentiating B cells.
![Repression of Erg by TFAP4 in MYC+ developing B cells is required for tumor suppression. Expression of Erg in pretransformed B220+ IgM− pro/pre-B cells from Eμ-Myc Tfap4+/+ mice and Eμ-Myc Tfap4+/− mice (A) and pro/pre-B tumor cells from Eμ-Myc Tfap4+/+, Eμ-Myc Tfap4+/−, and Eμ-Myc Tfap4−/− mice (B), as determined by RNA sequencing. (C) Colocalization of TFAP4 binding and accessible chromatin in the Erg locus (red vertical lines indicate exons; black horizontal lines represent introns) in developing B cells, pre-B cells (B220+ IgM−), pro/pre-B cells (CD19+); Fr.BC (CD19+ IgM− CD43+ CD24+), and Fr.D (CD19+ IgM− CD43− B220+). Y-axes show tag counts in 107 mapped reads. The Gene Expression Omnibus accession number for each dataset is indicated. (D) Expression of Tfap4, Myc, and Erg mRNA in developing B cells from the Immunological Genome Project database. (E) Sorting strategy for MYC+ and MYC− pro-B cells in Myc+/MYC-GFP protein reporter mice (far left, near left, and near right panels).21 Expression of Erg mRNA in MYC+ vs MYC− pro-B cells (far right panel). mRNA expression measured by quantitative reverse transcription polymerase chain reaction was normalized to Hprt1. (F) Expression of Erg in MYC+ pro-B cells from Tfap4+/+, Tfap4+/−, and Tfap4−/− mice harboring the MYC-GFP reporter. (G) Expression of Erg in CD19+ IgM– pro/pre-B cells from Eμ-Myc–Tfap4+/+ (non-Tg), Eμ-Myc Tfap4+/+, Tfap4+/− and Tfap4−/− mice prior to tumor development. (H) Survival of Tfap4-haploinsufficient Eμ-Myc mice with restricted Erg expression (Cd79aicre/+ [n = 20], median survival, 131 days; ErgF/+ Cd79aicre/+ [n = 8], median survival, 93.5 days; Tfap4F/+ ErgF/+ Cd79aicre/+ [n = 17], median survival, 94 days; Tfap4F/+ Cd79aicre/+ [n = 9], median survival, 62 days). (I) Frequencies of immature (Rag1 and/or VpreB1–expressing and surface Ig−) and mature (Rag1 and VpreB1–nonexpressing and surface Ig− or Ig+) B-cell tumors in Eμ-Myc mice with the indicated genotypes (Eμ-Myc Tfap4F/+Cd79aicre/+, n = 3; Eμ-Myc Tfap4F/+ErgF/+Cd79aicre, n = 6). *P < .05, **P < .01, ***P < .001, ****P < .0001, unpaired t test (A,E), one-way ANOVA with Tukey’s post hoc test (B,F-G), log-rank test, adjusted for multiple comparisons (H). ATAC-seq, assay for transposase-accessible chromatin with sequencing; ChIP-seq, chromatin immunoprecipitation sequencing; Fr., fraction; Imm, immature; Lg, large; ns, no statistical significance; RNA-seq, RNA sequencing; RPKM, reads per kilobase of transcript, per million mapped reads; Sm, small; TSS, transcription start site; α-, anti-.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/24/10.1182_blood.2021011711/5/m_bloodbld2021011711f5.png?Expires=1769079988&Signature=HFaJu8luBB3UfDFpULqCjxXu5vUEonYZbBG3bNkem9Ntl8CibTnLYZx2iKHj-R~YiFq4flHc8ofwSBJfFhMdsLmtmajWwb1MBkv5bqoGa0-9JTxGVfF-CcDHoNZyGIOmLer2FNMB8M2BLsdZxUTi8ycFpzyah-Ut~1jE7IDImpRdstayI-9Bt~VW0O2LzlWGRBLtgixH5wP8ZjeKpiPP687Q1IVExF7JUoFSpnO3DeNhFJrXIRhqJPyJKtcdAPbX6gdslzAtCgW6LB8vZ0O9ItE73WkRknyCgISwRObAEssGauwNpmusxZwzm4dstTNZpYllNuJ0xoHt45yqZDyAOQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Repression of Erg by TFAP4 in MYC+ developing B cells is required for tumor suppression. Expression of Erg in pretransformed B220+ IgM− pro/pre-B cells from Eμ-Myc Tfap4+/+ mice and Eμ-Myc Tfap4+/− mice (A) and pro/pre-B tumor cells from Eμ-Myc Tfap4+/+, Eμ-Myc Tfap4+/−, and Eμ-Myc Tfap4−/− mice (B), as determined by RNA sequencing. (C) Colocalization of TFAP4 binding and accessible chromatin in the Erg locus (red vertical lines indicate exons; black horizontal lines represent introns) in developing B cells, pre-B cells (B220+ IgM−), pro/pre-B cells (CD19+); Fr.BC (CD19+ IgM− CD43+ CD24+), and Fr.D (CD19+ IgM− CD43− B220+). Y-axes show tag counts in 107 mapped reads. The Gene Expression Omnibus accession number for each dataset is indicated. (D) Expression of Tfap4, Myc, and Erg mRNA in developing B cells from the Immunological Genome Project database. (E) Sorting strategy for MYC+ and MYC− pro-B cells in Myc+/MYC-GFP protein reporter mice (far left, near left, and near right panels).21 Expression of Erg mRNA in MYC+ vs MYC− pro-B cells (far right panel). mRNA expression measured by quantitative reverse transcription polymerase chain reaction was normalized to Hprt1. (F) Expression of Erg in MYC+ pro-B cells from Tfap4+/+, Tfap4+/−, and Tfap4−/− mice harboring the MYC-GFP reporter. (G) Expression of Erg in CD19+ IgM– pro/pre-B cells from Eμ-Myc–Tfap4+/+ (non-Tg), Eμ-Myc Tfap4+/+, Tfap4+/− and Tfap4−/− mice prior to tumor development. (H) Survival of Tfap4-haploinsufficient Eμ-Myc mice with restricted Erg expression (Cd79aicre/+ [n = 20], median survival, 131 days; ErgF/+ Cd79aicre/+ [n = 8], median survival, 93.5 days; Tfap4F/+ ErgF/+ Cd79aicre/+ [n = 17], median survival, 94 days; Tfap4F/+ Cd79aicre/+ [n = 9], median survival, 62 days). (I) Frequencies of immature (Rag1 and/or VpreB1–expressing and surface Ig−) and mature (Rag1 and VpreB1–nonexpressing and surface Ig− or Ig+) B-cell tumors in Eμ-Myc mice with the indicated genotypes (Eμ-Myc Tfap4F/+Cd79aicre/+, n = 3; Eμ-Myc Tfap4F/+ErgF/+Cd79aicre, n = 6). *P < .05, **P < .01, ***P < .001, ****P < .0001, unpaired t test (A,E), one-way ANOVA with Tukey’s post hoc test (B,F-G), log-rank test, adjusted for multiple comparisons (H). ATAC-seq, assay for transposase-accessible chromatin with sequencing; ChIP-seq, chromatin immunoprecipitation sequencing; Fr., fraction; Imm, immature; Lg, large; ns, no statistical significance; RNA-seq, RNA sequencing; RPKM, reads per kilobase of transcript, per million mapped reads; Sm, small; TSS, transcription start site; α-, anti-.
Repression of Erg by TFAP4 in MYC+ developing B cells is required for tumor suppression. Expression of Erg in pretransformed B220+ IgM− pro/pre-B cells from Eμ-Myc Tfap4+/+ mice and Eμ-Myc Tfap4+/− mice (A) and pro/pre-B tumor cells from Eμ-Myc Tfap4+/+, Eμ-Myc Tfap4+/−, and Eμ-Myc Tfap4−/− mice (B), as determined by RNA sequencing. (C) Colocalization of TFAP4 binding and accessible chromatin in the Erg locus (red vertical lines indicate exons; black horizontal lines represent introns) in developing B cells, pre-B cells (B220+ IgM−), pro/pre-B cells (CD19+); Fr.BC (CD19+ IgM− CD43+ CD24+), and Fr.D (CD19+ IgM− CD43− B220+). Y-axes show tag counts in 107 mapped reads. The Gene Expression Omnibus accession number for each dataset is indicated. (D) Expression of Tfap4, Myc, and Erg mRNA in developing B cells from the Immunological Genome Project database. (E) Sorting strategy for MYC+ and MYC− pro-B cells in Myc+/MYC-GFP protein reporter mice (far left, near left, and near right panels).21 Expression of Erg mRNA in MYC+ vs MYC− pro-B cells (far right panel). mRNA expression measured by quantitative reverse transcription polymerase chain reaction was normalized to Hprt1. (F) Expression of Erg in MYC+ pro-B cells from Tfap4+/+, Tfap4+/−, and Tfap4−/− mice harboring the MYC-GFP reporter. (G) Expression of Erg in CD19+ IgM– pro/pre-B cells from Eμ-Myc–Tfap4+/+ (non-Tg), Eμ-Myc Tfap4+/+, Tfap4+/− and Tfap4−/− mice prior to tumor development. (H) Survival of Tfap4-haploinsufficient Eμ-Myc mice with restricted Erg expression (Cd79aicre/+ [n = 20], median survival, 131 days; ErgF/+ Cd79aicre/+ [n = 8], median survival, 93.5 days; Tfap4F/+ ErgF/+ Cd79aicre/+ [n = 17], median survival, 94 days; Tfap4F/+ Cd79aicre/+ [n = 9], median survival, 62 days). (I) Frequencies of immature (Rag1 and/or VpreB1–expressing and surface Ig−) and mature (Rag1 and VpreB1–nonexpressing and surface Ig− or Ig+) B-cell tumors in Eμ-Myc mice with the indicated genotypes (Eμ-Myc Tfap4F/+Cd79aicre/+, n = 3; Eμ-Myc Tfap4F/+ErgF/+Cd79aicre, n = 6). *P < .05, **P < .01, ***P < .001, ****P < .0001, unpaired t test (A,E), one-way ANOVA with Tukey’s post hoc test (B,F-G), log-rank test, adjusted for multiple comparisons (H). ATAC-seq, assay for transposase-accessible chromatin with sequencing; ChIP-seq, chromatin immunoprecipitation sequencing; Fr., fraction; Imm, immature; Lg, large; ns, no statistical significance; RNA-seq, RNA sequencing; RPKM, reads per kilobase of transcript, per million mapped reads; Sm, small; TSS, transcription start site; α-, anti-.
To directly determine whether Erg derepression accelerates tumorigenesis, we deleted one Erg allele in Eμ-Myc Tfap4+/− mice, because complete loss of Erg arrests B-cell development.52Erg haploinsufficiency significantly delayed tumor onset and extended the survival of Eμ-Myc Tfap4+/− mice (Figure 5H). Furthermore, B-cell tumors that developed in Erg+/− mice were skewed toward the mature phenotype lacking expression of Rag1 and Vpreb1 (Figure 5I), indicating prolonged latency to transformation after MYC was turned on. These results indicate that TFAP4 suppresses the MYC-mediated transformation of B-cell precursors, at least in part, through the regulation of Erg.
TFAP4 suppresses tumorigenesis by coupling c-MYC–driven proliferation and differentiation of B-cell progenitors
ERG maintains the undifferentiated state of HSCs by inhibiting c-MYC targets, whereas c-MYC antagonizes self-renewal in favor of differentiation.41,53 Because our data demonstrated that c-MYC suppresses Erg through TFAP4, we hypothesized that TFAP4 prevents B-cell progenitors from maintaining their undifferentiated state or stemness while rapidly proliferating, thus restricting transformation. To test this, we adoptively transferred a mixture of Tfap4+/+ and Tfap4+/− CD19+ cells transduced with c-MYC marked by distinct reporters, with comparable infection efficiency across genotypes, into sublethally irradiated congenic mice and assessed their expansion and differentiation (Figure 6A; supplemental Figure 6H). Three weeks after transfer, Tfap4+/− cells became dominant among the total transduced donor-derived B220+ cells (Figure 6B-C). In addition to superior expansion and persistence of MYC+Tfap4+/− cells, frequencies of IgM+ cells were significantly lower in Tfap4+/− cells compared with Tfap4+/+ cells (Figure 6D). These results imply that Tfap4+/− pro/pre-B cells maintain their undifferentiated state during c-MYC–driven proliferation as a result of high Erg expression. Indeed, reducing Erg expression to 50% in Tfap4+/− pro/pre-B cells diminished the expansion of overall donor-derived cells, primarily as a result of the reduced expansion of Tfap4+/− cells relative to Tfap4+/+ cells. Reduction of Erg in Tfap4+/+ cells did not have any additional effect on the expansion of transferred cells (Figure 6C,E). Also, reducing Erg expression rescued the differentiation of transferred Tfap4+/− MYC+ pro/pre-B cells, as determined by the frequencies of IgM+ cells (Figure 6F).
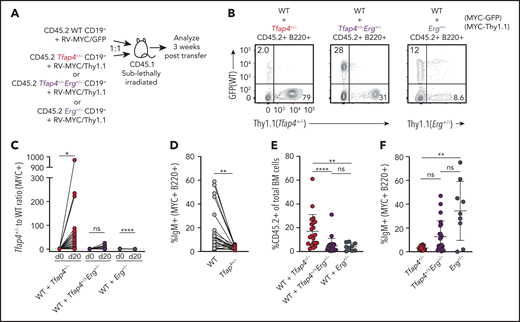
TFAP4-mediated restriction of Erg is required for coupling of c-MYC–dependent proliferation and differentiation. (A) Scheme for in vivo assessment of expansion and differentiation of Tfap4+/+, Tfap4+/−, Tfap4+/−Erg+/−, and Erg+/− CD19+ pro/pre-B cells transduced with a distinct retrovirus encoding mouse c-MYC (T58A). MSCV-based retroviruses expressing mouse MYC-ires-GFP and mouse MYC-ires-Thy1.1 were packaged by transiently transfecting PlatE cells with a helper plasmid, pCL-10A1. Viral supernatant harvested 48 hours after transfection was used to infect freshly isolated CD45.2 CD19+ BM cells from 6- to 14-week-old mice (age matched within the experimental groups) with the indicated genotypes at 1000g, 30°C for 1.5 to 2 hours in the presence of 10 mg/mL Polybrene. Transduced CD19+ BM cells from Tfap4+/+ (WT) and Tfap4+/− or Tfap4+/−Erg+/− or Erg+/− mice were cotransferred into sublethally irradiated CD45.1 recipients at a 1:1 ratio. Comparable infection efficiency across genotypes when using MYC-GFP or MYC-Thy1.1 retroviruses was achieved, as shown in supplemental Figure 6H. Three weeks later, mice were euthanized, and the proportions of each transduced cell population and their differentiation status were analyzed. (B) Representative flow cytometry plots showing frequencies of Tfap4+/+ (WT) and Tfap4+/− (left panel), WT and Tfap4+/−Erg+/− (middle panel), and WT and Erg+/− (right panel) donor-derived B220+ cells in the BM of recipient mice 20 days after transfer. (C) Ratios of c-MYC–expressing Tfap4+/− to WT, Tfap4+/−Erg+/− to WT, and Erg+/− to WT cells in the BM of recipient mice 20 days after transfer. (D) Percentages of surface IgM+ transduced Tfap4+/+ B220+ cells compared with Tfap4+/− B220+ cells in the same recipient mouse. (E) Percentages of CD45.2+ cells in the BM of mice in (A). (F) Percentages of surface IgM+ transduced Tfap4+/− B220+ cells compared with Tfap4+/−Erg+/− or Erg+/− B220+ cells (WT + Tfap4+/−, n = 21; WT + Tfap4+/−Erg+/−, n = 23; WT + Erg+/−, n = 9), Data are from 3 independent experiments combined. *P < .05, **P < .01, ****P < .0001; paired Student t test (C-D), **P < .01, ****P < .0001; Kruskal -Wallis test with Dunn’s multiple-comparison test (E,F). ns, no statistical significance.
TFAP4-mediated restriction of Erg is required for coupling of c-MYC–dependent proliferation and differentiation. (A) Scheme for in vivo assessment of expansion and differentiation of Tfap4+/+, Tfap4+/−, Tfap4+/−Erg+/−, and Erg+/− CD19+ pro/pre-B cells transduced with a distinct retrovirus encoding mouse c-MYC (T58A). MSCV-based retroviruses expressing mouse MYC-ires-GFP and mouse MYC-ires-Thy1.1 were packaged by transiently transfecting PlatE cells with a helper plasmid, pCL-10A1. Viral supernatant harvested 48 hours after transfection was used to infect freshly isolated CD45.2 CD19+ BM cells from 6- to 14-week-old mice (age matched within the experimental groups) with the indicated genotypes at 1000g, 30°C for 1.5 to 2 hours in the presence of 10 mg/mL Polybrene. Transduced CD19+ BM cells from Tfap4+/+ (WT) and Tfap4+/− or Tfap4+/−Erg+/− or Erg+/− mice were cotransferred into sublethally irradiated CD45.1 recipients at a 1:1 ratio. Comparable infection efficiency across genotypes when using MYC-GFP or MYC-Thy1.1 retroviruses was achieved, as shown in supplemental Figure 6H. Three weeks later, mice were euthanized, and the proportions of each transduced cell population and their differentiation status were analyzed. (B) Representative flow cytometry plots showing frequencies of Tfap4+/+ (WT) and Tfap4+/− (left panel), WT and Tfap4+/−Erg+/− (middle panel), and WT and Erg+/− (right panel) donor-derived B220+ cells in the BM of recipient mice 20 days after transfer. (C) Ratios of c-MYC–expressing Tfap4+/− to WT, Tfap4+/−Erg+/− to WT, and Erg+/− to WT cells in the BM of recipient mice 20 days after transfer. (D) Percentages of surface IgM+ transduced Tfap4+/+ B220+ cells compared with Tfap4+/− B220+ cells in the same recipient mouse. (E) Percentages of CD45.2+ cells in the BM of mice in (A). (F) Percentages of surface IgM+ transduced Tfap4+/− B220+ cells compared with Tfap4+/−Erg+/− or Erg+/− B220+ cells (WT + Tfap4+/−, n = 21; WT + Tfap4+/−Erg+/−, n = 23; WT + Erg+/−, n = 9), Data are from 3 independent experiments combined. *P < .05, **P < .01, ****P < .0001; paired Student t test (C-D), **P < .01, ****P < .0001; Kruskal -Wallis test with Dunn’s multiple-comparison test (E,F). ns, no statistical significance.
TFAP4 is a positive prognosis factor in MYC-high B-ALL
So far, our data have demonstrated that TFAP4 is mutated in human B-cell malignancies, and reduced TFAP4 functionality predisposes proliferating B cells to transformation in preclinical models. To gain insights into the role of TFAP4 in c-MYC–driven B-cell malignancies in humans, we reanalyzed the expression of c-MYC and TFAP4 in human pediatric B-ALL samples from the TARGET Phase II study (Figure 7A). Comparing patients with the top 25% and bottom 25% TFAP4/MYC ratios did not reveal a difference in the overall survival (data not shown). However, when we specifically compared samples with high MYC expression (z-score > 2), overall survival of patients with high TFAP4 expression was better than that of patients with low TFAP4 expression (Figure 7B-C). There was no difference in relative ERG expression between the 2 groups, possibly reflecting the selection of ERG-high clones during disease progression. Alternatively, the result also suggests an additional ERG-independent control of ALL progression by TFAP4 or ERG regulation by other oncogenic pathways (Figure 7D). These results suggest that upregulation of TFAP4 by c-MYC is a decisive factor for predisposition to B-cell malignancies, as well as is associated with the prognosis for patients with B-ALL.
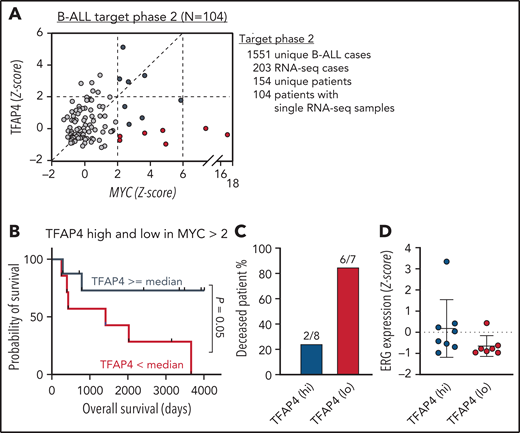
TFAP4 is a positive prognosis factor in B-ALL. (A) Plot showing normalized MYC and TFAP4 expression from 104 B-ALL patient samples in the TARGET phase 2 study. (B-C) Survival analysis of deceased patients with high vs low TFAP4 expression among patients with high MYC expression (z score >2; TFAP4lo median survival, 1413 days; TFAP4hi median survival, undefined). (D) Normalized ERG expression from B-ALL patient samples in (B). RNA-seq, RNA sequencing.
TFAP4 is a positive prognosis factor in B-ALL. (A) Plot showing normalized MYC and TFAP4 expression from 104 B-ALL patient samples in the TARGET phase 2 study. (B-C) Survival analysis of deceased patients with high vs low TFAP4 expression among patients with high MYC expression (z score >2; TFAP4lo median survival, 1413 days; TFAP4hi median survival, undefined). (D) Normalized ERG expression from B-ALL patient samples in (B). RNA-seq, RNA sequencing.
Discussion
Our study provides compelling evidence that c-MYC engages a tumor-suppressive program in B cells by inducing its direct downstream factor TFAP4. Coding mutations of TFAP4 prevail in lymphoid malignancies, particularly in a significant proportion of cases of BL. Our mouse models reveal substantial impacts on tumor onset by deletion or reduced expression of Tfap4 in developing B cells and mature B cells in the face of aberrant Myc expression. The tumor suppression is mediated, at least in part, by restricting the stemness factor ERG in immature B cells, because the reduced expression of Erg was sufficient to rescue developing B cells from aberrant expansion and accelerated transformation.
During hematopoiesis, HSCs give rise to various mature subsets through defined pathways. Because several proto-oncogenes are essential for normal hematopoiesis, their expression levels and timing must be tightly regulated to restrict the risk of leukemogenesis. Nonetheless, clonal hematopoiesis carrying oncogenic translocations is found in healthy individuals at a higher incidence than that of corresponding leukemias, including the t(8;14) translocation modeled by Eμ-Myc mice.54-56 Therefore, appropriate engagement of tumor suppression is essential to prevent these clones from transformation. HSCs and undifferentiated progenitors generate greater numbers of mature progenies through proliferation that is tightly linked to differentiation and at the cost of their self-renewing potential. Thus, perturbation of normal differentiation could result in a decoupling of the loss of self-renewal capacity and proliferation, which has been associated with hematopoietic malignancies.57-63 In HSCs, the antagonistic interplay between c-MYC and ERG is essential for balancing self-renewal and differentiation.41 ERG is essential for self-renewal of undifferentiated HSCs through suppression of c-MYC target genes; accordingly, loss of ERG closely recapitulates c-MYC overexpression in HSCs, resulting in depletion of undifferentiated HSCs.41,53 We demonstrate that the balance between MYC and ERG controls self-renewal and differentiation of B-cell progenitors. Insufficient TFAP4 induction and subsequent Erg persistence selectively impair the differentiation program by c-MYC, allowing c-MYC–expressing cells to continue proliferation with an undifferentiated phenotype and eventually form tumors. Although a subset of B-ALL is characterized by reduced ERG activity that is likely caused by an independent mechanism, elevated ERG was noted in a majority of cases of B-ALL that also express elevated c-MYC.
The frequency of TFAP4 mutations is rare in human B-ALL. However, within MYC-high B-ALL cases, the level of TFAP4 expression is associated with patient prognosis. Because increased TFAP4 expression is associated with poor prognosis in some cancers,64-66 lymphocytes may have established unique gene expression circuitry that converts the c-MYC–TFAP4 axis from a proproliferative/tumorigenesis module into a tumor-suppressive module by restricting their distinct targets. Similar counterregulation of MYC-driven proliferation may also be involved in the suppression of lymphomagenesis in GCB cells. GCB cells retain high proliferative potential similar to pro/pre-B cells, and TFAP4 may analogously function as a tumor suppressor by tightly coupling the expression of MYC and the restriction of continued proliferation. Thus, the induction of TFAP4 by MYC impacts the predisposition of B-cell malignancies, as well as likely contributes to disease progression. A therapeutic benefit for B-cell malignancies marked by high MYC expression and low TFAP4 could be achieved by inhibiting SCF ubiquitin ligase, leading to general cell cycle inhibition and specific stabilization of TFAP4, thus increasing the level of TFAP4 and balancing MYC and TFAP4 levels.67,68 We uncover that perturbation of the c-MYC–initiated and TFAP4-mediated feedback mechanism dramatically accelerates tumorigenesis.
Acknowledgments
The authors thank Vivek Durai, Sunnie Hsiung, Tenzin Yangdon, Jason Walker, and Mike White for technical support; Barry Sleckman for MYC-GFP mice; Kenneth Murphy, Eugene Oltz, Jacqueline Payton, and Dan Littman for discussions and critical reading of the manuscript; the Maxim Artyomov laboratory for the development of the RNA sequencing analysis tool Phantasus; and the Genome Technology Access Center (Department of Genetics, Washington University School of Medicine) for help with genomic analyses.
This work was supported by National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases grants R56 AI114593-01A1 (T.E.) and R01 AI130152-01A1 (T.E.), and NIH, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) grant R01 AR046000 (M.I.); Leukemia and Lymphoma Society Scholar Award 1349-18 (T.E.); a Siteman Investment Program Research Development Award (T.E.); and a Hu and Zeng Predoctoral Scholarship (E.T.). The Washington University Rheumatic Diseases Research Resource-based Center is supported by NIH, NIAMS grant P30 AR073752. The Genome Technology Access Center is supported, in part, by NIH, National Cancer Institute Cancer Center Support Grant P30CA91842 to the Siteman Cancer Center, and by the Institute of Clinical and Translational Sciences/Clinical and Translational Science Award (UL1TR000448) from the National Center for Research Resources, a component of the NIH, and the NIH Roadmap for Medical Research.
Authorship
Contribution: T.E. conceived the project; E.T., C.C., and T.E. designed experiments and interpreted the results; E.T., Y.T., C.C., Y.X., M.H., C.F., S.R., and T.E. performed experiments; G.S.C. helped with the analysis of whole-exome sequencing data; M.I. provided Erg-flox mice; and E.T. and T.E. wrote the manuscript with editorial comments from all of the other authors.
Conflict-of-interest disclosure: T.E. and C.C. have filed a provisional US patent application to propose the use of stabilized TFAP4 to improve T-cell function (Application Number 16343951 “AP4 and Methods of Promoting T Cell Activation”). The remaining authors declare no competing financial interests.
Correspondence: Takeshi Egawa, Washington University in St Louis, 660 South Euclid Ave, Campus Box 8118, St. Louis, MO 63110; e-mail: egawat@wustl.edu.
The sequencing data reported in this article have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus and Sequence Read Archive (accession numbers GSE133514 and PRJNA551263).
Data sharing requests should be sent to Takeshi Egawa (egawat@wustl.eu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal