

Key Points
ANP32B loss impairs the function of normal and CML stem cells.
ANP32B inhibits p53 activity to regulate hematopoiesis and CML leukemogenesis.

Abstract
Proper regulation of p53 signaling is critical for the maintenance of hematopoietic stem cells (HSCs) and leukemic stem cells (LSCs). The hematopoietic cell–specific mechanisms regulating p53 activity remain largely unknown. Here, we demonstrate that conditional deletion of acidic leucine-rich nuclear phosphoprotein 32B (ANP32B) in hematopoietic cells impairs repopulation capacity and postinjury regeneration of HSCs. Mechanistically, ANP32B forms a repressive complex with p53 and thus inhibits the transcriptional activity of p53 in hematopoietic cells, and p53 deletion rescues the functional defect in Anp32b-deficient HSCs. Of great interest, ANP32B is highly expressed in leukemic cells from patients with chronic myelogenous leukemia (CML). Anp32b deletion enhances p53 transcriptional activity to impair LSC function in a murine CML model and exhibits synergistic therapeutic effects with tyrosine kinase inhibitors in inhibiting CML propagation. In summary, our findings provide a novel strategy to enhance p53 activity in LSCs by inhibiting ANP32B and identify ANP32B as a potential therapeutic target in treating CML.
Introduction
Hematopoietic stem cells (HSCs) sustain production of multiple blood lineages throughout their lifetime, whereas leukemia stem cells (LSCs) are responsible for initiation and propagation of leukemia and significantly related to therapy failure and recurrence of the malignancies.1-4 Chronic myelogenous leukemia (CML) results from malignant transformation of HSCs by the BCR-ABL1 oncogene, which is generated by t(9;22) chromosome translocation.5 Tyrosine kinase inhibitors (TKIs) are highly effective in inducing remission of patients with CML, but they fail to target LSCs of CML, and recurrence is commonly seen following discontinuation of TKI treatment.5-7 Several self-renewal regulators have been investigated in both HSC and LSC contexts.8,9 The tumor suppressor p53, a well-known transcriptional regulator during the cellular stress response, is a critical regulator of HSCs behavior and essential to maintain HSCs quiescence through targeting Gfi-1 and Necdin10-12 and acts as the mediator of enhanced apoptosis of LSCs.13-15 Dual targeting of p53 and c-MYC was also shown selectively to eliminate LSCs from patients with CML.16 Despite the widely recognized role of p53 in HSCs and LSCs maintenance, the understanding of hematopoietic-specific mechanisms to regulate p53 activity remains limited.
ANP32B, a member of the acidic leucine-rich nuclear phosphoprotein 32 kDa (ANP32) family of proteins, is characterized by an N-terminal leucine-rich repeat (LRR) domain and a C-terminal low-complexity acidic region (LCAR).17 Constitutive Anp32b knockout in the pure C57BL/6 background is perinatal lethal.18 Mechanistically, ANP32B binds to transcription factors as a histone chaperone and modulates their activities.19,20 It is also required for the nucleocytoplasmic transport of specific mRNAs.21,22 We also reported that ANP32B is a direct substrate for caspase-3 and serves as a master enforcer of cell proliferation in breast cancer cells.23,24 However, the expression and function of ANP32B in normal hematopoiesis and leukemogenesis remain largely unknown.
Here, we investigate the contribution of ANP32B to normal and CML stem cell function using hematopoietic-specific Anp32b knockout mice and demonstrate that ANP32B plays a pivotal role in controlling normal and CML stem cell maintenance by inhibiting p53 activity.
Methods
Mouse competitive reconstitution analyses
Bone marrow (BM) cells or long-term (LT)-HSCs from mouse CD45.2 donor were mixed with CD45.1 competitor BM cells followed by intravenous injection into 8- to 10-week-old CD45.1 mice preconditioned with lethal irradiation. Secondary transplantation was performed with total BM cells from primary recipients. Percentages of donor-derived peripheral blood (PB) and BM cells were analyzed at the indicated times.
CML model and imatinib therapy
Ten- to 12-week-old donor mice were injected with 5-fluorouracil (5-FU) at 150 mg/kg body weight via tail vein. On day 6 after injection, Lin− BM cells were sorted and infected twice with BCR-ABL1-internal ribosome entry site(IRES)-green fluorescent protein (GFP) retrovirus. After the second round of infection, cells were collected and IV injected into lethally irradiated C57BL/6 mice. For imatinib therapy, the drug was administered by oral gavage twice a day at a dose of 100 mg/kg.
Quantification and statistical analysis
For comparison between 2 experimental groups (different genotypes or different treatments) or a specific pair within a group of multiple, a 2-tailed, unpaired Student t test was used, and error bars denote mean ± standard error of the mean (SEM). For survival of 5-FU challenge mice and CML recipients, statistical significance between different genetic and/or treatment conditions was assessed using the log-rank test. For comparison of proliferation curves for KU812 cells, 2-way analysis of variance was used. Differences were considered significant at P < 0.05.
Results
Deletion of Anp32b reduces HSC numbers by promoting quiescence
We assessed the mRNA levels of Anp32b in various hematopoietic populations in BM of C57BL/6 mice and revealed that ANP32B was highly expressed in HSCs, including LT-HSCs, short-term (ST)-HSCs, multipotent progenitors (MPPs), common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs), and common lymphoid progenitors (CLPs) in comparison with mature lymphoid, myeloid, and erythroid lineages (Figure 1A; supplemental Figure 1A-D available on the Blood Web site). Thus, we assessed the potential roles of ANP32B in hematopoiesis. To this end, we generated Anp32bfl/fl mice and crossed them with Scl-Cre transgenic mice, which drives deletion of Anp32b in HSCs, to produce Scl-Cre;Anp32bfl/fl mice (supplemental Figure 1E). Full deletion of Anp32b was observed 21 days after tamoxifen treatment (Figure 1B). Hereafter, tamoxifen-treated Scl-Cre−; Anp32bfl/fl and Scl-Cre+;Anp32bfl/fl mice are referred as Anp32b+/+ and Anp32b−/− mice, respectively. The ablation of ANP32B protein was confirmed by immunoblotting in Lin–, Lin+, and total BM cells of these mice. As expected, ANP32B expression was physiologically higher in Lin– cells than Lin+ cells (Figure 1C). Of great interest, the frequencies and absolute cell numbers of Lin–Sca-1+c-Kit+ (LSK) cells, including LT-HSCs, ST-HSCs, and MPPs, were significantly lower in Anp32b−/− mice compared with Anp32b+/+ mice at 4 weeks after tamoxifen induction (Figure 1D-F). This observation was also confirmed when the SLAM markers CD48 and CD150 were used to define HSCs (supplemental Figure 1F-G).25 However, frequencies of Lin–Sca-1–Kit+ (LK) progenitor cells including CMPs, GMPs, MEPs, and CLPs remained unchanged (Figure 1G-H). Meanwhile, the numbers of Anp32b-null primitive myeloid progenitor cells (colony forming unit-granulocyte, erythrocyte, monocyte and megakaryocyte, CFU-GEMM colonies) and differentiated myeloid progenitor cells (colony forming unit-granulocyte and macrophage, CFU-GM colonies) were mildly decreased, but those of erythroid progenitor cells (colony forming unit-erythrocyte, CFU-E colonies) were increased to an extent on Anp32b deletion compared with wild-type (WT) counterparts (supplemental Figure 1H). No difference was detected in the distribution of BM lymphoid, myeloid, and erythroid lineages between Anp32b−/− and Anp32b+/+ mice (Figure 1I), and the total BM cell number in Anp32b−/− mice was not changed 4 weeks after tamoxifen treatment (Figure 1J). We also monitored hematopoietic compartments in the BM of Anp32b−/− mice for 1 year after tamoxifen treatment and found remarkable decreases in the number of LT-HSCs, ST-HSCs, and MPPs together with only mild decreases in CMPs, MEPs, CLPs, and total BM, but no leukemia (supplemental Figure 1I-M).

Deletion of Anp32b reduces HSC numbers by promoting quiescence. (A) Relative Anp32b mRNA levels were measured by quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) in mouse BM LT-HSCs (Lin−Sca-1+c-Kit+Flk2−CD34−), ST-HSCs (Lin−Sca-1+c-Kit+Flk2−CD34+), MPPs (Lin−Sca-1+c-Kit+Flk2+), CLPs (Lin−Sca-1lowc-KitlowCD16/32+Flk2+), CMPs (Lin−Sca-1−c-Kit+CD16/32−CD34+), MEPs (Lin−Sca-1−c-Kit+CD16/32−CD34−), GMPs (Lin−Sca-1−c-Kit+CD16/32+CD34+), myeloid cells (Mac-1+/Gr-1+), B cells (B220+), and erythroid cells (TER119+) (n = 3). (B) Anp32b deletion was evaluated by genotyping in peripheral blood mononuclear cells (PBMCs) from Scl-Cre−;Anp32bfl/fl and Scl-Cre+;Anp32bfl/fl mice at indicated times after tamoxifen treatment. (C) ANP32B deletion was evaluated by Western blot in total BM cells and Lin+ and Lin− cells from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice. (D-F) Representative fluorescence-activated cell sorter (FACS) profiles (D), frequencies (E), and absolute cell numbers (F) of LSK cells (Lin−Sca-1+c-Kit+), LT-HSCs, ST-HSCs, and MPPs in BM from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice (n = 7). (G-I) Frequencies of LK (Lin−Sca-1−c-Kit+), CMPs, GMPs, MEPs (G), CLPs (H), and erythroid cells, myeloid cells, T cells (CD3E+), and B cells (I) in BM from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice (n = 7). (J) Total number of BM cells was calculated in 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice (n = 11). (K-L) Cell-cycle analysis of LSK cells (K) and LT-HSCs (L) in BM from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice (n = 4). Representative FACS profiles (i) and percentages of cell cycle distributions (ii) are shown. (M) Cell division tracing of LT-HSCs sorted from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice. LT-HSCs were stained with CFSE and cultured for 4 days. Cell divisions were multicolored, and the number of cell divisions is shown. Percentages of cells in each generation were calculated (n = 3). G, generation. (N) Proliferation assay of LSK cells and LT-HSCs in Anp32b+/+ and Anp32b−/− mice. Mice were intraperitoneally injected with BrdU and fed with BrdU-containing drinking water for 2 days. Representative FACS plots (i) and the statistical analysis of BrdU-positive cells in LSK cells and LT-HSCs (ii) (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed, unpaired Student t test (E-N). All animal experiments were repeated at least twice with similar results, and the results of 1 representative experiment are shown.
Deletion of Anp32b reduces HSC numbers by promoting quiescence. (A) Relative Anp32b mRNA levels were measured by quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) in mouse BM LT-HSCs (Lin−Sca-1+c-Kit+Flk2−CD34−), ST-HSCs (Lin−Sca-1+c-Kit+Flk2−CD34+), MPPs (Lin−Sca-1+c-Kit+Flk2+), CLPs (Lin−Sca-1lowc-KitlowCD16/32+Flk2+), CMPs (Lin−Sca-1−c-Kit+CD16/32−CD34+), MEPs (Lin−Sca-1−c-Kit+CD16/32−CD34−), GMPs (Lin−Sca-1−c-Kit+CD16/32+CD34+), myeloid cells (Mac-1+/Gr-1+), B cells (B220+), and erythroid cells (TER119+) (n = 3). (B) Anp32b deletion was evaluated by genotyping in peripheral blood mononuclear cells (PBMCs) from Scl-Cre−;Anp32bfl/fl and Scl-Cre+;Anp32bfl/fl mice at indicated times after tamoxifen treatment. (C) ANP32B deletion was evaluated by Western blot in total BM cells and Lin+ and Lin− cells from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice. (D-F) Representative fluorescence-activated cell sorter (FACS) profiles (D), frequencies (E), and absolute cell numbers (F) of LSK cells (Lin−Sca-1+c-Kit+), LT-HSCs, ST-HSCs, and MPPs in BM from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice (n = 7). (G-I) Frequencies of LK (Lin−Sca-1−c-Kit+), CMPs, GMPs, MEPs (G), CLPs (H), and erythroid cells, myeloid cells, T cells (CD3E+), and B cells (I) in BM from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice (n = 7). (J) Total number of BM cells was calculated in 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice (n = 11). (K-L) Cell-cycle analysis of LSK cells (K) and LT-HSCs (L) in BM from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice (n = 4). Representative FACS profiles (i) and percentages of cell cycle distributions (ii) are shown. (M) Cell division tracing of LT-HSCs sorted from 8- to 10-week-old Anp32b+/+ and Anp32b−/− mice. LT-HSCs were stained with CFSE and cultured for 4 days. Cell divisions were multicolored, and the number of cell divisions is shown. Percentages of cells in each generation were calculated (n = 3). G, generation. (N) Proliferation assay of LSK cells and LT-HSCs in Anp32b+/+ and Anp32b−/− mice. Mice were intraperitoneally injected with BrdU and fed with BrdU-containing drinking water for 2 days. Representative FACS plots (i) and the statistical analysis of BrdU-positive cells in LSK cells and LT-HSCs (ii) (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed, unpaired Student t test (E-N). All animal experiments were repeated at least twice with similar results, and the results of 1 representative experiment are shown.
To address how HSCs frequency was decreased in Anp32b−/− HSCs, we examined the cell cycle of HSCs by using Hoechst 33342 and Ki67 staining and found that approximately 59% and 91% of LSK cells and LT-HSCs, respectively, were in G0in Anp32b−/− mice compared with 49% and 85%, respectively, in Anp32b+/+ mice (Figure 1K-L). In vitro carboxyfluorescein diacetate succinimidyl ester (CFSE) tracing showed that Anp32b−/− LT-HSCs divided more slowly than Anp32b+/+ LT-HSCs (Figure 1M). In vivo bromodeoxyuridine (BrdU) incorporation labeling also showed the relatively lower proliferation rate of Anp32b−/− LSK cells and LT-HSCs (Figure 1N). These results suggest that Anp32b−/− HSCs are more quiescent and less likely to divide and proliferate. In contrast, we did not observe a significant difference in apoptosis between Anp32b+/+ and Anp32b−/− LSK cells and LT-HSCs (supplemental Figure 1N-O). Overall, our data suggest that Anp32b deletion in HSCs prevents proliferation by maintaining HSCs quiescence.
Anp32b deficiency reduces long-term repopulation capacity of HSCs
To test the long-term self-renewal capacity of HSCs from Anp32b−/− mice, we transplanted BM cells from Anp32b+/+ or Anp32b−/− mice with CD45.1 competitor cells into lethally irradiated recipients. We found that repopulation by Anp32b−/− BM cells was significantly lower compared with WT cells at 4, 8, 12, and 16 weeks after transplant (Figure 2A). Anp32b−/− LSK cells, including LT-HSCs, ST-HSCs, and MPPs, also exhibited a dramatic reduction at 16 weeks after transplant (Figure 2B). We did not observe any biased lineage reconstitution (Figure 2C), implying that ANP32B does not affect HSCs differentiation. On transplantation into secondary recipients, donor cell reconstitution of Anp32b−/− cells was significantly reduced compared with Anp32b+/+ cells (Figure 2D). Considering the notable decrease in HSC frequencies in Anp32b−/− mice, we performed another competitive repopulation experiment with the same number of LT-HSCs from Anp32b+/+ or Anp32b−/− mice. Similar results were observed after primary (Figure 2E) and secondary transplantations (Figure 2F). To exclude the possibility that the decreased repopulation was caused by defect of homing in Anp32b−/− HSCs, CFSE-labeled Anp32b+/+ or Anp32b−/− BM cells were injected into lethally irradiated recipients, followed by detection of CFSE+ and CFSE+ LSK cells in the BM, spleens, and livers. No significant differences were detected between Anp32b+/+ and Anp32b−/− donors at 16 hours after transplant (supplemental Figure 2A-B). In addition, BM cells from Scl-Cre−;Anp32bfl/fl and Scl-Cre+;Anp32bfl/fl mice were transplanted into WT recipients. At week 8 after transplant, recipient mice were injected with tamoxifen to induce Anp32b deletion. PB chimerism analysis confirmed the results (supplemental Figure 2C). Together, these data indicate that Anp32b deletion reduces long-term repopulation capacity of HSCs.
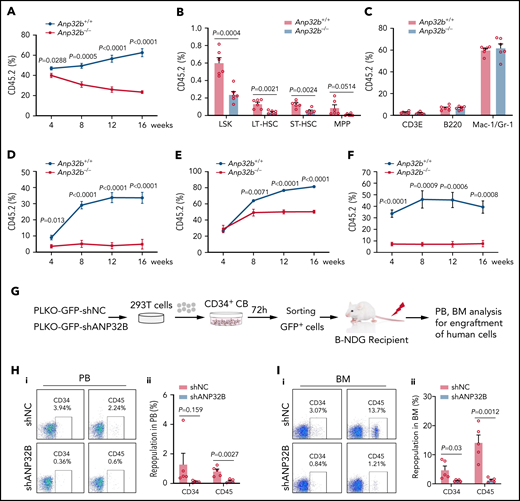
Anp32b deficiency reduces long-term repopulation capacity of HSCs. (A) Primary competitive transplantation assay was conducted with Anp32b+/+ and Anp32b−/− BM CD45.2 cells (6 × 105) along with 6 × 105 BM cells from CD45.1 competitor. Percentages of donor-derived cells in PB were analyzed at the indicated time points (n = 6). (B-C) Frequencies of donor-derived LSK cells, LT-HSCs, ST-HSCs, and MPPs (B) and lineage cells (C) in BM were analyzed at 16 weeks after transplant (n = 6). (D) Secondary transplantation was performed with total BM cells (1 × 106) from primary recipients in panel A at 16 weeks after transplant. Percentages of donor-derived cells in PB were analyzed at the indicated time points (n = 6). (E-F) Primary transplantation was conducted with FACS purified LT-HSCs (2 × 103) from Anp32b+/+ and Anp32b−/− BM cells along with 6 × 105 BM cells from CD45.1 competitor (E, n = 6). Secondary transplantation was performed with total BM cells (1 × 106) from primary recipients at 16 weeks after transplant (F; n = 5). Percentages of donor-derived cells in PB were analyzed at the indicated time points. (G) Schematic strategy of evaluation of the in vivo effect of ANP32B knockdown in human cord blood CD34+ cells. (H-I) Representative FACS plots (i) and percentages (ii) of human CD34+ and CD45+ cells engrafted in PB cells (H) and BM cells (I) 6 weeks after transplantation (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed unpaired Student t test (A-F, H-I). All animal experiments were repeated at least twice with similar results, and the results of 1 representative experiment are shown.
Anp32b deficiency reduces long-term repopulation capacity of HSCs. (A) Primary competitive transplantation assay was conducted with Anp32b+/+ and Anp32b−/− BM CD45.2 cells (6 × 105) along with 6 × 105 BM cells from CD45.1 competitor. Percentages of donor-derived cells in PB were analyzed at the indicated time points (n = 6). (B-C) Frequencies of donor-derived LSK cells, LT-HSCs, ST-HSCs, and MPPs (B) and lineage cells (C) in BM were analyzed at 16 weeks after transplant (n = 6). (D) Secondary transplantation was performed with total BM cells (1 × 106) from primary recipients in panel A at 16 weeks after transplant. Percentages of donor-derived cells in PB were analyzed at the indicated time points (n = 6). (E-F) Primary transplantation was conducted with FACS purified LT-HSCs (2 × 103) from Anp32b+/+ and Anp32b−/− BM cells along with 6 × 105 BM cells from CD45.1 competitor (E, n = 6). Secondary transplantation was performed with total BM cells (1 × 106) from primary recipients at 16 weeks after transplant (F; n = 5). Percentages of donor-derived cells in PB were analyzed at the indicated time points. (G) Schematic strategy of evaluation of the in vivo effect of ANP32B knockdown in human cord blood CD34+ cells. (H-I) Representative FACS plots (i) and percentages (ii) of human CD34+ and CD45+ cells engrafted in PB cells (H) and BM cells (I) 6 weeks after transplantation (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed unpaired Student t test (A-F, H-I). All animal experiments were repeated at least twice with similar results, and the results of 1 representative experiment are shown.
Considering that there were no differences in the distribution of lymphoid, myeloid, and erythroid lineages in BM between Anp32b−/− and Anp32b+/+ mice and there were only mild decreases in CMPs, MEPs, and CLPs in Anp32b−/− mice maintained for 1 year regardless of remarkable decreases in the numbers of LT-HSCs, ST-HSCs, and MPPs (supplemental Figure 1I-L), we monitored the effects of ANP32B ablation on normal hematopoiesis in conditions of stress hematopoiesis. For this purpose, we treated Anp32b+/+ and Anp32b−/− mice with 5-FU and found that at day 18 after 5-FU treatment, the numbers of LSK cells (including LT-HSCs, ST-HSCs, and MPPs) and progenitor cells (including CMPs, GMPs, MEPs, and CLPs) were dramatically reduced in Anp32b−/− mice compared with controls (supplemental Figure 2D-F). Meanwhile, the percentage of HSCs in G0 phase was markedly increased in Anp32b−/− mice compared with controls (supplemental Figure 2G). CFSE tracing also showed that the LT-HSC division was significantly slower in Anp32b−/− mice than Anp32b+/+ mice after 5-FU treatment (supplemental Figure 2H). Especially, sequential 5-FU challenge assay revealed that Anp32b−/− mice died dramatically earlier than Anp32b+/+ mice (supplemental Figure 2I). These results indicate that the effects of Anp32b ablation on normal hematopoiesis are more apparent in conditions of stress hematopoiesis.
To address the function of ANP32B in human HSCs, we used shRNA to specifically knockdown ANP32B in CD34+ cells from human cord blood (supplemental Figure 2J). Following ANP32B knockdown, human CD34+ cells grew much more slowly (supplemental Figure 2K) and formed significantly fewer colonies (supplemental Figure 2L). When human CD34+ cells with ANP32B knockdown were transplanted into NDG-SCID mice, the engraftment of human CD45+ and CD34+ cells was dramatically decreased in PB and BM in comparison with control human CD34+ cells (Figure 2G-I), suggesting that ANP32B loss also inhibits human HSCs function. Together, these results indicate that ANP32B is essential for maintaining long-term repopulating activity of human HSCs.
ANP32B directly interacts with p53
As a histone chaperone, ANP32B has been shown to be involved in binding to transcription factors and regulating transcription.19,20 To explore the potential ANP32B-interacting proteins in HSCs, we transfected empty or Flag-tagged ANP32B plasmids into 32D cells (a mouse myeloid progenitor cell line), and cell lysates were immunoprecipitated by anti-Flag antibody. The precipitates were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by in-gel digestion and liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis. In total, we identified 62 proteins shared in 2 independent experiments, including p53. Pathway analysis using Gene Ontology indicated that ANP32B-interacting proteins are significantly involved in p53 signaling in addition to transcriptional regulation in general (supplemental Figure 3A; supplemental Table 3). Validating these results, Flag-ANP32B pulled down p53 in 32D cells (Figure 3A), and Flag-p53 pulled down ANP32B (supplemental Figure 3B). Endogenous ANP32B–p53 interaction was also seen in 32D and mouse LSK cells (Figure 3B-C). Furthermore, in vitro interaction assay showed that either GST-tagged or S-tagged p53 pulled down His-tagged ANP32B (Figure 3D; supplemental Figure 3C), supporting a direct interaction of ANP32B with p53 protein.
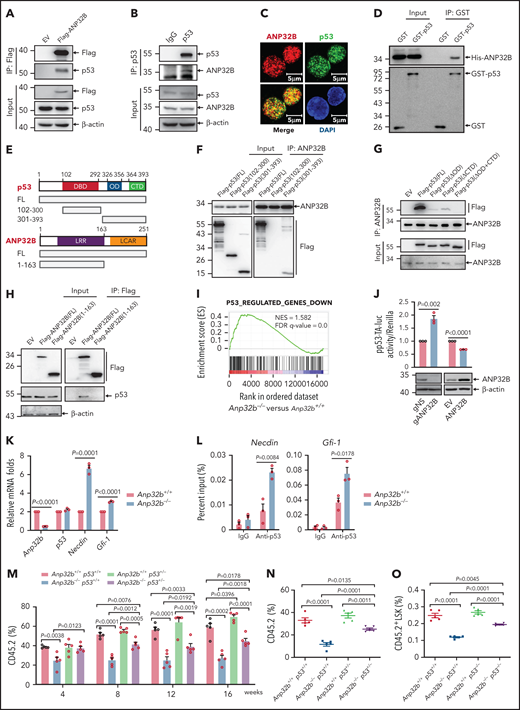
ANP32B interacts with and inhibits the transcriptional activity of p53 to maintain the function of HSCs. (A) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of Flag-tagged ANP32B-transfected 32D cells. (B) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of endogenous p53 in 32D cells. (C) Immunofluorescent staining of endogenous ANP32B, p53 together with restaining of 4′,6-diamidino-2-phenylindole in mouse LSK cells, followed by imaging with confocal microscopy. (D) Bacterially expressed His-ANP32B was incubated with GST or GST-tagged p53, followed by GST-tag pulldown and Western blot analysis of indicated proteins. (E) Structure schematic diagram of full-length and truncated segments of p53 and ANP32B. (F-G) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of anti-ANP32B antibody in H1299 cells transfected with Flag-p53 full-length plasmid and truncated segments. (H) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of anti-FLAG M2 beads in 293T cells transfected with Flag-ANP32B full-length plasmid and N163 segments. (I) GSEA analysis of RNA-seq data from Anp32b+/+ and Anp32b−/− LSK cells using p53-regulated gene set (n = 3 biologically independent p53+/+ and p53−/− HSPCs obtained from the GEO, accession code GSE137126). (J) Clonally derived HCT116 cell lines depleted of ANP32B (gANP32B) or not (gNS), empty vector (EV), or ANP32B-infected HCT116 cells were cotransfected with a luciferase reporter plasmid for p53 transcription (pp53-TA-luc) and Renilla luciferase reporter plasimid, and the relative luciferase activity were determined. (K) Relative mRNA expression levels of indicated genes in BM LSK cells from Anp32b+/+ and Anp32b−/− mice by quantitative RT-PCR. (L) ChIP-quantitative RT-PCR of immunoglobulin G and p53 on the promoters of the indicated genes in BM LSK cells from Anp32b+/+ and Anp32b−/− mice. (M-O) Competitive transplantation assay was conducted with Anp32b+/+p53+/+, Anp32b−/−p53+/+, Anp32b+/+p53+/−, and Anp32b−/−p53+/− BM CD45.2 cells (6 × 105) along with 6 × 105 BM cells from CD45.1 competitor. Percentages of CD45.2+ cells in PB were analyzed at the indicated time points (M). Frequencies of CD45.2+ cells (N) and CD45.2+LSK cells (O) in BM were analyzed at 16 weeks after transplant (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed, unpaired Student t test (J-O). The experiments in panels A-D, F-H, and J were repeated 3 times independently with similar results, and the results of 1 representative experiment are shown. The animal experiments were repeated twice with similar results, and the results of 1 representative experiment are shown.
ANP32B interacts with and inhibits the transcriptional activity of p53 to maintain the function of HSCs. (A) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of Flag-tagged ANP32B-transfected 32D cells. (B) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of endogenous p53 in 32D cells. (C) Immunofluorescent staining of endogenous ANP32B, p53 together with restaining of 4′,6-diamidino-2-phenylindole in mouse LSK cells, followed by imaging with confocal microscopy. (D) Bacterially expressed His-ANP32B was incubated with GST or GST-tagged p53, followed by GST-tag pulldown and Western blot analysis of indicated proteins. (E) Structure schematic diagram of full-length and truncated segments of p53 and ANP32B. (F-G) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of anti-ANP32B antibody in H1299 cells transfected with Flag-p53 full-length plasmid and truncated segments. (H) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of anti-FLAG M2 beads in 293T cells transfected with Flag-ANP32B full-length plasmid and N163 segments. (I) GSEA analysis of RNA-seq data from Anp32b+/+ and Anp32b−/− LSK cells using p53-regulated gene set (n = 3 biologically independent p53+/+ and p53−/− HSPCs obtained from the GEO, accession code GSE137126). (J) Clonally derived HCT116 cell lines depleted of ANP32B (gANP32B) or not (gNS), empty vector (EV), or ANP32B-infected HCT116 cells were cotransfected with a luciferase reporter plasmid for p53 transcription (pp53-TA-luc) and Renilla luciferase reporter plasimid, and the relative luciferase activity were determined. (K) Relative mRNA expression levels of indicated genes in BM LSK cells from Anp32b+/+ and Anp32b−/− mice by quantitative RT-PCR. (L) ChIP-quantitative RT-PCR of immunoglobulin G and p53 on the promoters of the indicated genes in BM LSK cells from Anp32b+/+ and Anp32b−/− mice. (M-O) Competitive transplantation assay was conducted with Anp32b+/+p53+/+, Anp32b−/−p53+/+, Anp32b+/+p53+/−, and Anp32b−/−p53+/− BM CD45.2 cells (6 × 105) along with 6 × 105 BM cells from CD45.1 competitor. Percentages of CD45.2+ cells in PB were analyzed at the indicated time points (M). Frequencies of CD45.2+ cells (N) and CD45.2+LSK cells (O) in BM were analyzed at 16 weeks after transplant (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed, unpaired Student t test (J-O). The experiments in panels A-D, F-H, and J were repeated 3 times independently with similar results, and the results of 1 representative experiment are shown. The animal experiments were repeated twice with similar results, and the results of 1 representative experiment are shown.
To map the domains of ANP32B and p53 required for their interactions, Flag-tagged full-length, DNA-binding domain (aa102-300) and the 301-393 fragment of p53 (Figure 3E) were transfected into p53-deficient H1299 cells, followed by co-immunoprecipitation with ANP32B antibody. As depicted in Figure 3F, ANP32B pulled down full-length and the 301-393 fragment but not the DNA-binding domain of p53, suggesting that the C-terminal moiety of p53 is essential for its interaction with ANP32B. Next, cells were transfected with a Flag-tagged oligomerization domain (OD, aa326-356) deletion mutant and/or an unstructured C-terminal domain (CTD, aa364-393) deleted mutants (ΔOD, ΔCTD, ΔOD+CTD) of p53,26 followed by immunoprecipitation with anti-ANP32B antibody. In accordance, ΔOD+CTD mutant of p53 failed to interact with ANP32B (Figure 3G). Additionally, the N-terminal (aa1-163) of ANP32B did not interact with p53 (Figure 3H). Collectively, our data suggest that both OD and CTD of p53 are required for its interaction with the C-terminal acidic domain of ANP32B.
ANP32B inhibits the transcriptional activity of p53 in HSCs
After ruling out that Anp32b loss did not change mRNA and protein levels of p53 in LSK cells of BM (supplemental Figure 3D), we performed gene set enrichment analysis (GSEA) to gain a global view of the transcriptome profile regulated by ANP32B. Interestingly, p53 target genes were significantly enriched in the transcriptome of Anp32b knockout LSK cells (Figure 3I), suggesting that ANP32B may negatively regulate the transcriptional activity of p53. Hence, we examined the possible effect of ANP32B on the DNA binding activity of p53 by gel shift assay. As shown in supplemental Figure 3E, addition of His-ANP32B gradually eliminated the shift band of the p53-DNA probe complex, indicating that ANP32B possesses the ability to negatively regulate DNA-binding activity of p53. In line with this, a specific p53 responsive element–driven luciferase assay showed that the transcriptional activity of p53 was significantly increased on ANP32B knockdown and decreased on ANP32B overexpression (Figure 3J). Accordingly, Gfi-1 and Necdin, 2 well-known p53-target genes in HSCs,10-12 but not Bax, Puma, Bak1, and Tnfrsf10b,27-30 were upregulated in Anp32b−/− LSK cells (Figure 3K; supplemental Figure 3F). Furthermore, chromatin immunoprecipitation (ChIP) assays on endogenous p53 in Anp32b+/+ or Anp32b−/− LSK cells revealed that Anp32b deficiency enhanced binding of p53 to the promoters of Gfi-1 and Necdin genes (Figure 3L). Taken together, these data indicate that ANP32B acts as a transcriptional repressor of p53 in HSCs.
To detect whether p53 signaling mediates the functional defect in Anp32b−/− HSCs, we generated Anp32b−/− and p53+/− mice by crossing Scl-Cre;Anp32bfl/fl mice with p53+/− mice, followed by tamoxifen treatment to delete Anp32b. Competitive repopulation assays revealed partial rescue of the repopulation defect in the Anp32b−/−p53+/− mice compared with Anp32b−/−p53+/+ mice, evidenced by the increased percentage of donor-derived cells in PB and BM (Figure 3M-N), donor-derived LSK cells (Figure 3O), and BrdU+ LSK cells (supplemental Figure 3G). These data suggest that p53 signaling mediates, at least partially, effects of ANP32B on HSC maintenance.
Anp32b deletion suppresses CML progression and impairs the function of LSCs
Next, we investigated whether ANP32B also has a role in maintaining LSCs using a retroviral model of CML, a paradigmatic stem cell disorder that can be recapitulated in mice by retrovirally introducing the p210 form of BCR-ABL1 into cycling hematopoietic stem/progenitor cells, followed by transplantation into lethally irradiated mice (supplemental Figure 4A).31 The model is characterized by dramatically increased peripheral white blood cell (WBC) counts (supplemental Figure 4B), higher percentage of myeloid (GFP+Gr-1+) leukemia cells (supplemental Figure 4C), lung hemorrhaging, massive splenomegaly, and hepatomegaly (supplemental Figure 4D-E) on day 15 after transplant.
To study the role of ANP32B in CML development, Anp32b+/+ or Anp32b−/− donor BM cells transduced with GFP-BCR-ABL1 retrovirus were used to induce CML. Recipients of Anp32b−/− BM cells developed CML significantly slower than recipients of WT BM cells. The total WBC number was largely reduced in PB of Anp32b−/− CML mice (Figure 4A), and Anp32b deficiency significantly inhibited growth of BCR-ABL1–expressing myeloid (GFP+Gr-1+) leukemia cells in PB and BM of recipients (Figure 4B-C). In line with this, in serial transplantation experiments, recipients of Anp32b−/− cells had markedly increased survival (Figure 4D-E), and a decrease in the size and weight of lung, spleen, and liver (Figure 4F), with much less severe infiltration of myeloid leukemia cells compared with their WT counterparts (Figure 4G). Of note, Anp32b loss did not affect the transduction efficiency of the BCR-ABL1 retrovirus (supplemental Figure 4F) or the homing of BCR-ABL1–expressing cells to the BM after transplantation (supplemental Figure 4G-H). In addition, Anp32b deletion substantially prolonged the survival of CML mice driven by BCR-ABL1T315I (supplemental Figure 4I), a CML model resistant to TKIs.32 Together, these results demonstrate that ANP32B exerts a role in CML development.
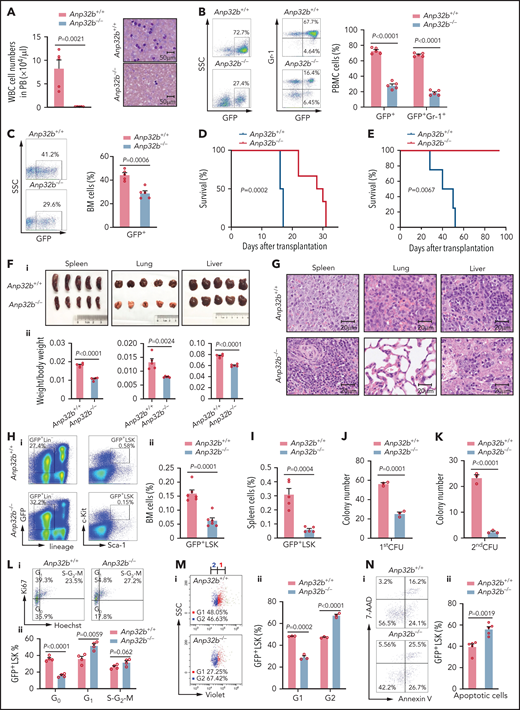
Loss of Anp32b impairs in vivo progression of CML and maintenance of LSCs. (A) Total number of WBCs in PB and Wright-Giemsa staining of PB smears were conducted 15 days after transplant from recipients received BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells (n = 5). (B-C) Representative FACS profiles and percentages of GFP+ cells and GFP+Gr-1+ cells in PB (B) and BM (C) 15 days after first transplantation (n = 5). (D) Survival curves for recipients transplanted with BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells (n = 6). (E) Survival curves for recipients receiving Anp32b+/+ and Anp32b−/− GFP+ leukemia cells (1 × 106) on secondary transplantation (n = 4). (F-G) Gross pathology (i) and relative weights (ii) (F) and hematoxylin-eosin staining (G) of the spleens, lungs, and livers from recipients received BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 15 days after transplant (n = 5). (H-I) FACS analysis of GFP+LSK cells in BM (H) or spleen (I) at 15 days after first transplantation. Representative FACS profiles (i) and the percentage of GFP+LSK cells (ii) are shown (n = 5). (J) Colony-forming assays were performed with BM GFP+LSK cells sorted from recipients receiving BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 12 days after transplant. Colony numbers were calculated at day 6 after plating (n = 3). (K) First plated leukemia cells were further collected and used for second plating at day 6. The numbers of colonies were calculated 7 days after plating (n = 3). (L) Cell cycle analysis of GFP+LSK cells in BM at 15 days after first transplantation Representative FACS profiles (i) and percentages of cell cycle distributions (ii) are shown (n = 4). (M) Cell division tracing of BM GFP+LSK cells. GFP+LSK cells were sorted from recipients received BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 15 days after transplant and stained with CellTrace Violet dye. After culturing for 24 hours, cell divisions were analyzed by flow cytometry, and the number of generations is shown. Percentages of cells in each generation were calculated (n = 3). G, generation. (N) Apoptosis analysis of GFP+LSK cells in BM 15 days after first transplantation. Representative FACS profiles (i) and the percentage of GFP+LSK cells (ii) are shown (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed, unpaired Student t test (A-C, F, H-N) or log-rank test (D-E). All animal experiments were repeated at least twice with similar results, and the results of 1 representative experiment are shown.
Loss of Anp32b impairs in vivo progression of CML and maintenance of LSCs. (A) Total number of WBCs in PB and Wright-Giemsa staining of PB smears were conducted 15 days after transplant from recipients received BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells (n = 5). (B-C) Representative FACS profiles and percentages of GFP+ cells and GFP+Gr-1+ cells in PB (B) and BM (C) 15 days after first transplantation (n = 5). (D) Survival curves for recipients transplanted with BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells (n = 6). (E) Survival curves for recipients receiving Anp32b+/+ and Anp32b−/− GFP+ leukemia cells (1 × 106) on secondary transplantation (n = 4). (F-G) Gross pathology (i) and relative weights (ii) (F) and hematoxylin-eosin staining (G) of the spleens, lungs, and livers from recipients received BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 15 days after transplant (n = 5). (H-I) FACS analysis of GFP+LSK cells in BM (H) or spleen (I) at 15 days after first transplantation. Representative FACS profiles (i) and the percentage of GFP+LSK cells (ii) are shown (n = 5). (J) Colony-forming assays were performed with BM GFP+LSK cells sorted from recipients receiving BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 12 days after transplant. Colony numbers were calculated at day 6 after plating (n = 3). (K) First plated leukemia cells were further collected and used for second plating at day 6. The numbers of colonies were calculated 7 days after plating (n = 3). (L) Cell cycle analysis of GFP+LSK cells in BM at 15 days after first transplantation Representative FACS profiles (i) and percentages of cell cycle distributions (ii) are shown (n = 4). (M) Cell division tracing of BM GFP+LSK cells. GFP+LSK cells were sorted from recipients received BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 15 days after transplant and stained with CellTrace Violet dye. After culturing for 24 hours, cell divisions were analyzed by flow cytometry, and the number of generations is shown. Percentages of cells in each generation were calculated (n = 3). G, generation. (N) Apoptosis analysis of GFP+LSK cells in BM 15 days after first transplantation. Representative FACS profiles (i) and the percentage of GFP+LSK cells (ii) are shown (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed, unpaired Student t test (A-C, F, H-N) or log-rank test (D-E). All animal experiments were repeated at least twice with similar results, and the results of 1 representative experiment are shown.
CML is a stem cell disorder in which disease initiation and maintenance rely on LSCs, a population of cells enriched in the Lin–Sca-1+c-Kit+ population.33 To assess the impact of Anp32b loss on LSCs from CML, we quantified the frequency and total cell number of LSCs (GFP+LSK cells) in control and Anp32b knockout CML mice. Anp32b deficiency caused a marked reduction of LSCs in either BM (Figure 4H) or spleen (Figure 4I). Furthermore, the number of colonies formed by Anp32b−/− CML LSK cells was significantly decreased compared with Anp32b+/+ controls in the first plating, and this impairment was even more impressive in the second plating (Figure 4J-K). Hoechst and Ki67 staining revealed a significantly higher percentage of cycling cells (Ki67high) accompanied by a decrease in G0 quiescent cells (HoechstlowKi67low) in Anp32b-null GFP+LSK cells (Figure 4L). In vitro CFSE tracing confirmed that GFP+LSK cells divided more rapidly in Anp32b KO mice than in WT mice (Figure 4M), suggesting that Anp32b deletion is sufficient to drive quiescent LSCs into cycling. Furthermore, the proportion of annexin-V+ apoptotic cells was markedly greater within Anp32b−/− GFP+LSK cells than within control GFP+LSK cells (Figure 4N). Collectively, Anp32b deficiency in LSCs results in enhanced proliferation and increased apoptosis.
p53 signaling rescues the Anp32b-deficiency CML phenotype
ANP32B inhibition did not change the BCR-ABL1 protein level and its activity in CML KU812 cells (supplemental Figure 5A) and GFP+ leukemia cells from mice (supplemental Figure 5B), suggesting that ANP32B regulates LSCs through a BCR-ABL1-independent mechanism. As p53 has an important role for CML LSCs survival,16 and ANP32B functions as a transcriptional repressor of p53 in HSCs, we hypothesized that p53 signaling also plays a role in ANP32B-dependent self-renewal of CML LSCs. The KU812 cell line was derived from a patient with CML in blast phase.34 The cells harbor a K132R p53 mutation, but this mutation is believed to not alter p53 function.35 As expected, the interaction between endogenous ANP32B and p53 was also found in KU812 cells by immunoblotting and immunofluorescence (Figure 5A-B). KU812 cells with ANP32B knockdown expanded much more slowly than control cells (Figure 5C-D). Consequently, p53 regulated genes were significantly enriched in transcriptome of KU812 cells with ANP32B knockdown (Figure 5E). Furthermore, several p53 target genes such as Bax, Puma, Bak1, and Tnfrsf10b,27-30 but not Gfi-1 and Necdin, were upregulated in Anp32b−/− CML LSK cells (Figure 5F), and Anp32b deficiency enhanced binding of p53 to the promoters of these genes in CML LSK cells (Figure 5G). All these results indicate that ANP32B also represses transcriptional activity of p53 in CML LSCs.
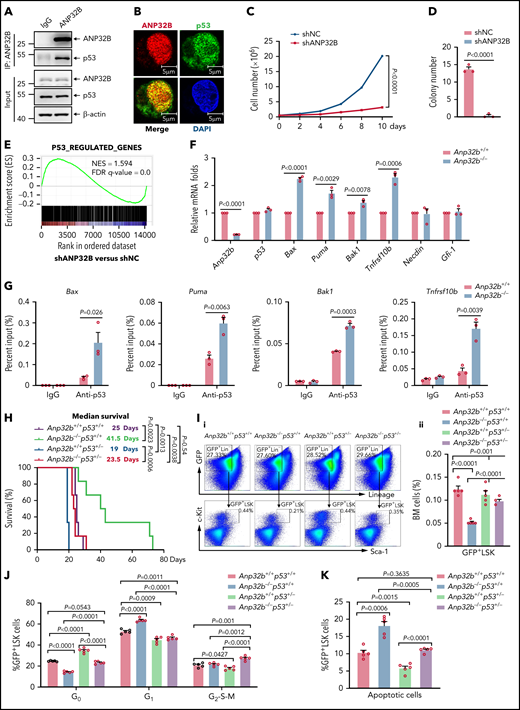
ANP32B-mediated transcriptional repression of p53 enables maintenance of CML LSCs. (A) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of endogenous ANP32B in KU812 cells. (B) Immunofluorescent staining of endogenous ANP32B and p53 together with restaining of 4′,6-diamidino-2-phenylindole in KU812 cells, followed by imaging with confocal microscopy. (C) Proliferation curves of KU812 cells infected with shNC or shANP32B. Cell numbers were counted at the indicated days (n = 3). (D) Colony-forming assay for KU812 cells infected with shNC or shANP32B. Colony numbers were evaluated at day 10 (n = 3). (E) GSEA analysis of RNA-seq data from KU812 cells with shNC and shANP32B infection using p53-regulated gene set (data obtained from CML CD34+ cells treated with RITA or not, fold change >2, false discovery rate (FDR) < 0.05, data were deposited in the European Nucleotide Archive under accession number PRJEB9942). (F) Relative mRNA expression levels of indicated genes in BM GFP+LSK cells sorted from recipients receiving BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 12 days after transplant by quantitative RT-PCR. (G) ChIP-quantitative RT-PCR of immunoglobulin G and p53 on the promoters of the indicated genes in BM GFP+LSK cells sorted from recipients receiving BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 12 days after transplant. (H) Survival curves and analysis of median survival from recipients transplanted with BCR-ABL1–transduced Anp32b+/+p53+/+, Anp32b+/+p53+/−, Anp32b−/−p53+/+, and Anp32b−/−p53+/− Lin− BM cells (n = 6). (I) Representative FACS plots (i) and the percentage (ii) of BM GFP+LSK cells at 14 days after transplant (n = 5). (J) Cell cycle analysis of BM GFP+LSK cells at 14 days after transplant. Percentages of cell cycle distributions are shown (n = 5). (K) Apoptosis analysis of BM GFP+LSK cells at 14 days after transplant. Percentage of apoptotic GFP+LSK cells are shown (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by 2-way analysis of variance (C), 2-tailed, unpaired Student t test (D,F-G,I-K), or log-rank test (H). The experiments in panels A-D were repeated 3 times independently with similar results, and the results of 1 representative experiment are shown. All animal experiments were repeated at least twice with similar results, and the results of 1 representative experiment are shown.
ANP32B-mediated transcriptional repression of p53 enables maintenance of CML LSCs. (A) Western blot analysis of indicated proteins in the inputs and immunoprecipitates of endogenous ANP32B in KU812 cells. (B) Immunofluorescent staining of endogenous ANP32B and p53 together with restaining of 4′,6-diamidino-2-phenylindole in KU812 cells, followed by imaging with confocal microscopy. (C) Proliferation curves of KU812 cells infected with shNC or shANP32B. Cell numbers were counted at the indicated days (n = 3). (D) Colony-forming assay for KU812 cells infected with shNC or shANP32B. Colony numbers were evaluated at day 10 (n = 3). (E) GSEA analysis of RNA-seq data from KU812 cells with shNC and shANP32B infection using p53-regulated gene set (data obtained from CML CD34+ cells treated with RITA or not, fold change >2, false discovery rate (FDR) < 0.05, data were deposited in the European Nucleotide Archive under accession number PRJEB9942). (F) Relative mRNA expression levels of indicated genes in BM GFP+LSK cells sorted from recipients receiving BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 12 days after transplant by quantitative RT-PCR. (G) ChIP-quantitative RT-PCR of immunoglobulin G and p53 on the promoters of the indicated genes in BM GFP+LSK cells sorted from recipients receiving BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells at 12 days after transplant. (H) Survival curves and analysis of median survival from recipients transplanted with BCR-ABL1–transduced Anp32b+/+p53+/+, Anp32b+/+p53+/−, Anp32b−/−p53+/+, and Anp32b−/−p53+/− Lin− BM cells (n = 6). (I) Representative FACS plots (i) and the percentage (ii) of BM GFP+LSK cells at 14 days after transplant (n = 5). (J) Cell cycle analysis of BM GFP+LSK cells at 14 days after transplant. Percentages of cell cycle distributions are shown (n = 5). (K) Apoptosis analysis of BM GFP+LSK cells at 14 days after transplant. Percentage of apoptotic GFP+LSK cells are shown (n = 5). Error bars denote mean ± SEM. Statistical significance was determined by 2-way analysis of variance (C), 2-tailed, unpaired Student t test (D,F-G,I-K), or log-rank test (H). The experiments in panels A-D were repeated 3 times independently with similar results, and the results of 1 representative experiment are shown. All animal experiments were repeated at least twice with similar results, and the results of 1 representative experiment are shown.
To validate the requirement of p53 signaling for ANP32B-mediated CML progression in vivo, Anp32b+/+p53+/+, Anp32b−/−p53+/+, Anp32b+/+p53+/−, and Anp32b−/−p53+/− mice were used to induce CML. The extended survival time (Figure 5H), decreased spleen, lung, and liver size and weight (supplemental Figure 5C), less severe infiltration with leukemia cells (supplemental Figure 5D), and reduced GFP+LSK cells (Figure 5I) in Anp32b−/− CML mice were greatly reversed in Anp32b−/−p53+/− CML mice. We further found that inactivation of p53 in LSCs resulted in increased quiescence and decreased apoptosis. As expected, inactivation of p53 in Anp32b−/− LSCs almost completely rescued impaired quiescence and increased apoptosis in Anp32b−/− LSCs (Figure 5J-K), suggesting that regulation of LSCs by ANP32B is predominantly mediated by p53 signaling.
ANP32B is highly expressed in patients with CML and maintains the long-term engraftment of human CML CD34+ cells
We compared ANP32B expression in BM from patients with CML and nonleukemia controls. Notably, ANP32B protein expression was significantly higher in CML BM cells than control ones (Figure 6A-B). To investigate whether targeting ANP32B might be clinically useful, we infected human CML CD34+ cells with lentiviruses carrying ANP32B shRNA or a scramble shRNA for 48 hours. We found that CML CD34+ cells formed significantly fewer colonies (Figure 6C; supplemental Figure 6A) on ANP32B knockdown. We also reintroduced shANP32B-resistant ANP32B into shANP32B-transduced CML CD34+ cells and found that re-expression of ANP32B could totally reverse ANP32B knockdown-induced inhibition of colony formation (Figure 6D-E), indicating the specificity of the shRNA to silence ANP32B. When these human CML CD34+ cells were transplanted into sublethally irradiated NDG mice (Figure 6F), engraftment of human CD34+, CD45+, and CD45+CD11b+ myeloid cells on ANP32B knockdown was reduced (Figure 6G-H). As genotyping was not performed, it is uncertain whether ANP32B knockdown reduced CML engraftment over residual normal cells.
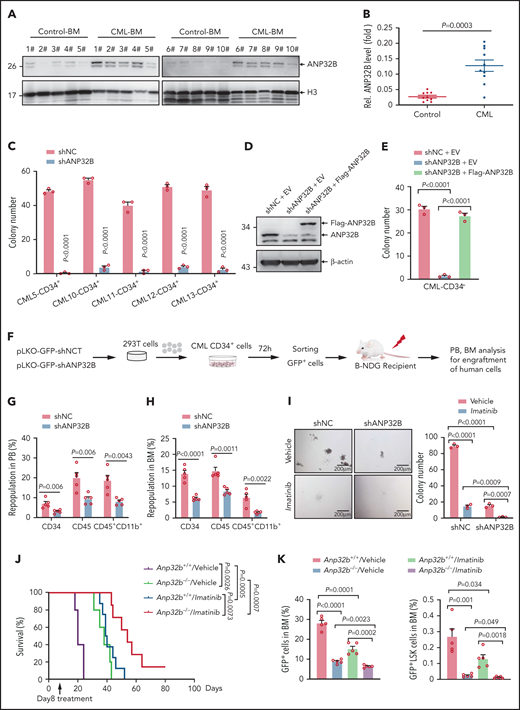
Targeting ANP32B impairs human CML development and synergizes with imatinib therapy to eradicate CML (A-B) Relative ANP32B protein expression levels in bone marrow mononuclear cells (BMMCs) of patients with CML and control patients were analyzed (n = 10). All samples were collected at diagnosis before therapy. All patients with CML were diagnosed in chronic phase and were BCR-ABL1 positive. (C) Colony-forming assay for human CML CD34+ cells infected with shNC or shANP32B. Numbers of colonies were evaluated at day 10 (n = 3). All samples were collected at diagnosis before therapy. All patients with CML were diagnosed in chronic phase and were BCR-ABL1 positive. (D) shNC- or shANP32B-infected human CML CD34+ cells were stably transfected with EV or Flag-tagged ANP32B with the shRNA target sequence mutation, and Western blots of indicated proteins are shown. (E) Colony-forming assay for shNC/EV, shANP32B/EV, and shANP32B/Flag-ANP32B CML CD34+ cells. Colony numbers were counted at day 10 after plating (n = 3). (F) Schematic strategy of evaluation of the in vivo effect of ANP32B knockdown in human CML CD34+ cells. (G-H) Percentages of human CD34+, CD45+, and CD45+CD11b+ cells engrafted in PB (G) and BM (H) 6 months after transplantation (n = 5). Human CD34+ cells sorted from CML patient 10 were used in this experiment. (I) Human CML CD34+ cells (CML patient 10) infected with shNC or shANP32B were seeded into methylcellulose medium with 5μM imatinib or dimethyl sulfoxide as vehicle. Colony numbers were counted at day 10 after plating (n = 3). (J-K) Survival curves for recipients transplanted with BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells followed by the treatment with either vehicle or imatinib at day 8 after transplantation (J, n = 5 for Anp32b+/+/vehicle and Anp32b−/−/vehicle group, n = 8 for Anp32b+/+/imatinib, and n = 7 for Anp32b−/−/imatinib group). Percentages of GFP+ cells and GFP+LSK cells in BM were analyzed at day 14 after transplantation (n = 5) (K) Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed, unpaired Student t test (B-C,E,G-I) or log-rank test (J). The experiments in panels J-K were repeated twice with similar results, and the results of 1 representative experiment are shown. The experiments in panels A-H are presented from an independent experiment.
Targeting ANP32B impairs human CML development and synergizes with imatinib therapy to eradicate CML (A-B) Relative ANP32B protein expression levels in bone marrow mononuclear cells (BMMCs) of patients with CML and control patients were analyzed (n = 10). All samples were collected at diagnosis before therapy. All patients with CML were diagnosed in chronic phase and were BCR-ABL1 positive. (C) Colony-forming assay for human CML CD34+ cells infected with shNC or shANP32B. Numbers of colonies were evaluated at day 10 (n = 3). All samples were collected at diagnosis before therapy. All patients with CML were diagnosed in chronic phase and were BCR-ABL1 positive. (D) shNC- or shANP32B-infected human CML CD34+ cells were stably transfected with EV or Flag-tagged ANP32B with the shRNA target sequence mutation, and Western blots of indicated proteins are shown. (E) Colony-forming assay for shNC/EV, shANP32B/EV, and shANP32B/Flag-ANP32B CML CD34+ cells. Colony numbers were counted at day 10 after plating (n = 3). (F) Schematic strategy of evaluation of the in vivo effect of ANP32B knockdown in human CML CD34+ cells. (G-H) Percentages of human CD34+, CD45+, and CD45+CD11b+ cells engrafted in PB (G) and BM (H) 6 months after transplantation (n = 5). Human CD34+ cells sorted from CML patient 10 were used in this experiment. (I) Human CML CD34+ cells (CML patient 10) infected with shNC or shANP32B were seeded into methylcellulose medium with 5μM imatinib or dimethyl sulfoxide as vehicle. Colony numbers were counted at day 10 after plating (n = 3). (J-K) Survival curves for recipients transplanted with BCR-ABL1–transduced Anp32b+/+ and Anp32b−/− Lin− BM cells followed by the treatment with either vehicle or imatinib at day 8 after transplantation (J, n = 5 for Anp32b+/+/vehicle and Anp32b−/−/vehicle group, n = 8 for Anp32b+/+/imatinib, and n = 7 for Anp32b−/−/imatinib group). Percentages of GFP+ cells and GFP+LSK cells in BM were analyzed at day 14 after transplantation (n = 5) (K) Error bars denote mean ± SEM. Statistical significance was determined by a 2-tailed, unpaired Student t test (B-C,E,G-I) or log-rank test (J). The experiments in panels J-K were repeated twice with similar results, and the results of 1 representative experiment are shown. The experiments in panels A-H are presented from an independent experiment.
Combining ANP32B ablation with imatinib or p53 activator promotes eradication of CML LSCs
BCR-ABL1 TKIs fail to eliminate LSCs in CML, which remains a potential source of relapse.6,7,36 Given that ANP32B is essential for maintenance of LSCs, we examined the effects of combining ANP32B ablation with imatinib. As shown in Figure 6I, ANP32B depletion in human CML CD34+ cells enhanced imatinib inhibition of colony formation. Moreover, ablation of ANP32B enhanced imatinib effects in vivo. As depicted in Figure 6J-K, mice treated with imatinib alone from day 8 after transplant showed extended survival and decreased BM GFP+LSK cells. Combination therapy with Anp32b ablation and imatinib markedly attenuated CML development and reduced BM GFP+LSK cells with concomitant reduction of spleen, liver, and lung pathology and size (supplemental Figure 6B-C). On the other hand, we treated human CML CD34+ cells with Nutlin-3a, the most extensively characterized inhibitor of the MDM2–p53 interaction and activity of which on cancer cells is dependent on p53 activation37,38 in combination with or without ANP32B knockdown. The results showed that, similar to ANP32B knockdown, Nutlin-3a dramatically inhibited colony formation by human CML CD34+ cells, which was further enhanced by ANP32B knockdown (supplemental Figure 6D).
Discussion
HSC quiescence is critical for preserving a lifelong pool of HSCs that can sustain a highly regenerative hematopoietic system. Quiescent HSCs are not often used in normal hematopoiesis and are most likely preserved for emergent hematopoiesis such as serial transplantation and postinjury regeneration.39-41 In this report, we demonstrate that ANP32B is a critical regulator of quiescence in HSCs in vivo. Although Anp32b deficiency did not cause severe BM defects either in adult or 1-year-old mice, loss of Anp32b prevented division and proliferation of HSCs and decreased frequency of adult Anp32b−/− HSCs compared with Anp32b+/+ controls during normal hematopoiesis. Competitive transplantation experiments revealed that Anp32b−/− HSCs showed a significantly lower level of donor cell reconstitution after primary transplantation, which was more pronounced after secondary transplantation. These results indicate that ANP32B exerts a role for maintaining the number and long-term repopulating capacity of HSCs. Especially, the effects on normal hematopoiesis induced by Anp32b ablation were more apparent in conditions of 5-FU–induced stress hematopoiesis, suggesting that ANP32B is more important for BM recovery after stress.
On the other hand, understanding how LSCs are regulated at a molecular level is crucial for eradicating leukemia by targeting LSCs. ANP32A, another member of ANP32 family, has been reported to be highly expressed in primary acute myeloid leukemia cells and promote leukemogenesis in vitro.42 Here we demonstrated that deletion of ANP32B attenuates BCR-ABL1 induced CML in vivo. Mounting evidence shows that p53 activity can be regulated by interacting with a series of cofactors.43-45 ANP32 family proteins are involved in regulating transcription and chromatin architecture.46 In line with this, we showed that ANP32B-interacting proteins are mainly involved in general transcriptional regulation. More interestingly, ANP32B directly interacts with p53 and acts as a corepressor of p53 to inhibit its DNA binding and transcriptional activity. Especially, impaired quiescence and increased apoptosis in Anp32b−/− LSCs were reversed in Anp32b−/−p53+/− LSCs, and inhibition of proliferation in Anp32b−/− HSCs was partially rescued in Anp32b−/−p53+/− HSCs, indicating that p53 is involved in the regulation of HSCs and LSCs by ANP32B.
As a transcription factor in charge of cell vital decisions, p53 controls the expression of several hundreds of genes, promoting cell fates ranging from cell cycle arrest to senescence and apoptosis, depending on the type of different stress.47-50 The p53-induced cell cycle arrest may occur at either G2/M or G1/S checkpoints, reflecting cell context–specific outcomes in different cells.47,50,51 Here, we found that Anp32b deletion in HSCs increased quiescent cells and impaired division and proliferation, whereas Anp32b deficiency in LSCs decreased quiescent cells and promoted their cell cycle entry, which was followed by enhanced proliferation and increased apoptosis, supporting that p53 plays different roles in fate decision of HSCs and LSCs as reviewed.52 Principally, p53 promotes quiescence of HSCs, and the absence of p53 promotes HSC more cell cycle entry, whereas p53 preferentially induces apoptosis of LSCs,13-16 although precise function of p53 on LSCs has not been clarified. Indeed, we showed that Anp32b−/− CML LSK cells presented upregulated expressions of p53 target genes Bax, Puma, Bak1, and Tnfrsf10b, which are involved in triggering LSC apoptosis.53 On the contrary, Anp32b−/− HSCs had increased expressions of Gfi-1 and Necdin, in which Gfi-1 has been shown to restrict HSC proliferation and preserve HSC functional integrity,54,55 and Necdin functions to maintain HSC quiescence.56 Further validation and understanding of the mechanisms through which p53 selectively regulates different genes in HSCs and LSCs deserves to be explored in the future.
The frequency of p53 mutations is relatively lower in leukemia than in other cancers.35,57,58 Therefore, fine-tuned regulation of p53 activity is critical to harness p53 activity for therapeutic purpose in leukemia. Intriguingly, ANP32B is highly expressed in cells from patients with CML, and the combination of ANP32B deficiency and imatinib therapy synergistically inhibited CML initiation and prolonged survival. Because regulation is mainly dependent on p53 in LSCs, but partially in HSCs by ANP32B, our observations provide a rationale for exploring ANP32B–p53 interaction inhibitors for elimination of LSCs without severely compromising the function of HSCs. In summary, our in vitro and in vivo results show that deletion or knockdown of ANP32B in HSCs and LSCs activate p53, impairing emergent hematopoiesis and CML leukemogenesis and supporting further investigation of ANP32B–p53 interaction inhibitors as an approach for targeting of CML LSCs in combination with TKIs.
Acknowledgments
The authors thank Dengli Hong and Kewen Zhao at Shanghai Jiao Tong University School of Medicine for kindly providing materials as described in the “Methods” section.
This work was supported by National Key Research and Development Program of China (grant 2020YFA0803403), National Natural Science Foundation (grants 91853206 and 81772936), and its innovative group support (grant 81721004), Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (grant 2019-I2M-5-051), and Shanghai Committee of Science and Technology (grant 20JC1410100).
Authorship
S.Y., X.-N.Z., and H.-L.Z. performed most experiments; Q.Y. and Y.-S.W. conducted partial experiments; D.Z., M.-D.L., and Y.-L.P. provided clinical samples; L.X. performed LC-MS/MS; P.H. and M.-K.G. performed bioinformatics analysis; S.-M.S., M.Z., Y.-L.W., and J.-K.Z. provided constructive comments and discussion; and Y.Y. and G.-Q.C. designed and supervised the entire project and prepared the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Guo-Qiang Chen; e-mail: chengq@shsmu.edu.cn; and Prof. Yun Yu yy@shsmu.edu.cn Rui-Jin hospital, Shanghai Jiao Tong University School of Medicine, No. 280 Chong Qing Rd. (S), Shanghai, 200025, China; e-mail: 2yy@shsmu.edu.cn.
The online version of this article contains a data supplement. For original data, please contact yy@shsmu.edu.cn

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal