


Abstract
Inborn errors of immunity (IEIs) are rare inherited disorders arising from monogenic germline mutations in genes that regulate the immune system. The majority of IEI are primary immunodeficiencies characterized by severe infection often associated with autoimmunity, autoinflammation, and/or malignancy. Allogeneic hematopoietic stem cell transplant (HSCT) has been the corrective treatment of choice for many IEIs presenting with severe disease in early childhood, and experience has made this a successful and comparatively safe treatment in affected children. Early HSCT outcomes in adults were poor, resulting in extremely limited use worldwide. This is changing because of a combination of improved IEI diagnosis to inform patient selection, better understanding of the natural history of specific IEI, and improvements in transplant practice. Recently published HSCT outcomes for adults with IEIs have been comparable with pediatric data, making HSCT an important option for correction of clinically severe IEIs in adulthood. Here we discuss our practice for patient selection, timing of HSCT, donor selection and conditioning, peri- and post-HSCT management, and our approach to long-term follow-up. We stress the importance of multidisciplinary involvement in the complex decision-making process that we believe is required for successful outcomes in this rapidly emerging area.
Introduction
Inborn errors of immunity (IEIs) encompass a large and heterogenous group of rare diseases caused by monogenic germline mutations in more than 400 separate genes that regulate the immune system.1 Most IEIs are primary immunodeficiency disorders characterized by severe and recurrent infection, which is associated with differing degrees of autoimmunity, autoinflammation, and malignancy secondary to dysregulated immunity. Enormous phenotypic variation is seen in IEIs, in part determined by the specific gene mutated and in part because of other factors, even within a single family, such as environment and epigenetic landscape.2-4 Although a monogenic diagnosis is frequently achieved when IEI presents in childhood, most IEI patients first presenting in adulthood do not yet have a known genetic diagnosis, even in sequenced cohorts.5
Hematopoietic stem cell transplant (HSCT) for severe pediatric onset IEI is well established and often the gold standard treatment. Indications for pediatric HSCT are agreed on by international groups such as the European Society for Blood and Marrow Transplantation (EBMT) and the American Society for Transplantation and Cellular Therapy (ASTCT) and are regularly revised to incorporate emerging data.6,7 In contrast, the indications for HSCT in adult IEI patients are less well described and have only recently been included in the updated EBMT and ASTCT indications for HSCT,6,7 as a clinical option for some diseases. Regularly updated guidelines are produced by European Society for Immunodeficiency (ESID)/EBMT Inborn Errors Working Party (IEWP) and the most recent version includes a section on adults with IEI.8
A rapidly growing body of publications detail HSCT outcomes for IEI in adults,9-25 and additional data on >200 adults have been published in abstract form. In most published series, the overall survival (OS) was similar to that achieved in children and infants (80% or greater at a median follow-up of between 14 months and 5 years). Age at transplantation (whether adolescent, young adult, or older adult) remains an important consideration, as several studies report less favorable outcomes (both OS and graft-versus-host disease [GVHD] rates) in older patients.9,10,14,19 The most frequent IEI diagnoses in published series including older adolescents and adult patients are chronic granulomatous disease (CGD), primary hemophagocytic lymphohistiocytosis (HLH) including genetically undefined HLH and GATA2 deficiency (including patients with associated bone marrow failure and/or hematologic malignancies in addition to immunodeficiency); however, in specialist centers, a much wider range of IEIs are proceeding to HSCT.
This review discusses how HSCT can be applied to adults with IEI and gives our own insights into how this can be done safely and for whom.
Patient selection: how we identify adult IEI patients who may benefit from HSCT
The potential benefit of HSCT should always outweigh the risks of the procedure. The challenge with many IEI patients that survive to, or present in, adulthood is that neither HSCT outcomes nor outcomes with conservative management are well described. Specific considerations in determining which adults should be transplanted are highlighted in Box 1.
Specific considerations in determining which adults should have HSCT
- •
Adult IEI patients being considered for HSCT are more likely to have had a mild phenotype in childhood or late onset presentation of an IEI-associated complication.
- •
The natural history of most IEI in adulthood are not well described and therefore the outlook for an individual patient with conservative management is often not easy to predict.
- •
The HSCT outcome data for some conditions are poor or unclear, for example, XIAP, CVID.
- •
We do not yet fully understand the pathogenesis and/or prognosis of some newly discovered genetic defects.
- •
Some diseases have extra hematopoietic aspects that may not be corrected by HSCT, for example, STAT3 LOF.
- •
Adults with IEIs generally have accumulated complications and end organ damage that increases risk or precludes HSCT.
- •
Fertility preservation for younger adults should include access to prenatal genetic diagnosis if indicated/requested.
To date, we have mainly reserved HSCT for adults with a monogenic IEI and severe clinical disease. Although the many different genetic causes and broad phenotypic variation present challenges for optimal patient selection, criteria have been agreed in the United Kingdom by an expert group26 (supplemental Table 1 available on the Blood Web site). To provide oversight for clinical practice and enable complex decision making around HSCT, all adult IEI patients in England being considered for HSCT are discussed at a national multidisciplinary meeting involving transplant physicians, immunologists, and other specialist physicians from multiple centers.
Case 1
Case 1 is a 42-year-old man with Wiskott-Aldrich syndrome (WAS). He has a known pathogenic mutation in the WAS gene resulting in preserved partial WAS protein (WASp) expression. He was diagnosed in infancy (positive family history) and his 2 older affected brothers both died of their disease: 1 in childhood from bleeding and 1 in adulthood from WAS-associated large vessel vasculitis. He had recurrent bacterial respiratory tract infections throughout life, requiring hospitalization, resulting in moderately severe bronchiectasis. He had recurrent mucosal bleeding related to severe thrombocytopenia (platelets < 10) and moderate eczema but no autoimmune or malignant complications. He was married with 2 young children and a full-time job. He was keen to explore HSCT given the relatively sudden deterioration and death of his brother in adulthood.
WAS is an X-linked IEI, characterized by microthrombocytopenia, recurrent infections, and eczema,27 commonly complicated by autoimmunity and hematologic malignancy that indicate severe disease.27,28 A milder form of WAS, known as X-linked thrombocytopenia (XLT),29 is typically associated with mutations that preserve partial protein expression and lack significant immunodeficiency or immune dysregulation,30 although complications can develop later in life.31 The use of HSCT for severe WAS is considered gold standard therapy in childhood with excellent outcomes,32 whereas patients with mild disease managed conservatively do well, and the use of HSCT for XLT is not routinely recommended.33
Case 2
Case 2 is a 24-year-old man with X-linked CGD. He was diagnosed following Aspergillus pneumonia at 17 years of age, which required an intensive care admission. Subsequently, he had an episode of severe pseudomonal pneumonia and developed CGD-associated colitis. He had progressive colitis after initial response to steroids, despite azathioprine and regular vedoluzimab infusions.
Cases 1 and 2 illustrate adult patients with monogenic IEIs diagnosed in childhood or adolescence but not transplanted who developed further disease complications in adulthood. HSCT may not have been offered earlier for various reasons including (i) satisfactory initial responses to conservative management and/or less severe phenotype; (ii) perceived or actual unacceptable risk of HSCT; (iii) HSCT not considered; or (iv) the formal IEI diagnosis was made later.
For case 1 (42 years, WAS), the presence of severe infections, bronchiectasis, and the need for immunoglobulin replacement therapy (IRT) place him in a moderately severe category. With optimal conservative management, the potential to develop additional autoimmune and malignant complications is high, suggesting his WAS may be life limiting with an accumulation of complications over an uncertain time frame.
For case 2 (24 years, X-CGD), his poorly controlled steroid refractory colitis and a previous history of severe fungal infection requiring intensive care unit admission predict a poor outcome with conservative therapy alone,34 which together with good outcomes following HSCT for adults with CGD,10,12 supported proceeding to HSCT in early adulthood.
Case 3
Case 3 is a 26-year-old woman. She was well until 19 years of age, when she developed vaccine-associated yellow fever requiring prolonged hospitalization. Subsequently human papillomavirus (HPV) warts affected her hands, feet, and perineum, leading to severe dysplasia and extensive anal intraepithelial neoplasia. She had subsequent inflammatory bowel disease and respiratory tract infections leading to bronchiectasis. She was treated with prophylactic antibiotics and IRT. Bone marrow examination confirmed trilineage myelodysplasia (MDS) with no increase in blasts. A subsequent genetic diagnosis of GATA2 deficiency was made.35,36
Case 3 typifies onset of IEI in adulthood with subsequent genetic diagnosis (newly described IEI). Achieving a genetic diagnosis assists HSCT decision making, particularly if the disease phenotype is only moderately severe or the disease is newly described with little or no HSCT experience. At the time, it was unclear whether the HPV-driven neoplasia would be amenable to HSCT. In this case, the presence of trilineage MDS (albeit with a low international prognostic scoring system score) and future risk of leukemic transformation supported the rationale for HSCT. Recent natural history data for GATA2 deficiency has confirmed poor outcomes with conservative management alone, with early death from infections and malignancy (35% death rate by 40 years of age).37 In contrast, good disease-free survival approaching 90% suggests HSCT may be the treatment of choice, although issues remain around optimal timing and risks of GVHD reported in some studies.21,38 For patients with bone marrow failure and/or leukemic transformation of MDS, the indication for HSCT is clear.37 The very recently reported lack of genotype–phenotype correlation39 adds further complexity to patient selection.
Case 4
Case 4 is a 35-year-old man with common variable immunodeficiency (CVID). He presented with progressive vitiligo and recurrent respiratory tract bacterial infections at 17 years of age. Pan-hypogammaglobulinemia was identified at 30 years of age, and he was diagnosed with CVID. Initial management was with prophylactic antibiotics and IRT. There was rapid development of multiple CVID-associated complications over 3 years: bronchiectasis, chronic norovirus gastrointestinal infection, splenomegaly, hypersplenism, granulomatous hepatitis with nodular regenerative hyperplasia on liver biopsy, and portal hypertension. Synthetic liver function was preserved. Grade 2 esophageal varices, spironolactone-controlled ascites, refractory norovirus infection, and progressively worsening quality of life were noted at his last review.
CVID is a clinical diagnosis and the most common form of immunodeficiency requiring treatment in adults.40 Most patients remain well on IRT with a normal life expectancy, but a subset with additional features of immune dysregulation develop progressive severe organ complications.41 Although this complex group is more likely to have a monogenic diagnosis, most remain genetically undefined even in thoroughly sequenced cohorts.5,42,43 The transition from being well on IRT to severely unwell with complex complications can occur rapidly and unpredictably. Patients may therefore present to transplant physicians too late with complications precluding HSCT. Published retrospective data for HSCT in adult patients with complex CVID have indicated worse outcomes than for other IEIs,14 underlining the need for better information to select patients and timing of HSCT in CVID. We do not currently recommend HSCT for these patients in the absence of a clinical study.
Optimal timing of HSCT in adult IEI patients
Many published studies have demonstrated worse outcomes with increasing age at HSCT, with a greater risk of GVHD and poorer OS.10,25,44,45 In pediatric series, typically age > 5 years at HSCT is associated with poorer outcomes, and in series including adolescents and adults, age at HSCT remains significant.10 However, recent detailed analysis of 284 patients ≥15 years of age at HSCT for IEI conducted by the EBMT IEWP has shown that number of IEI-related complications, comorbidity score before HSCT, and prior splenectomy influenced outcome rather than age (Albert MA, Wehr C, Suarez F, Fox ML, Gennery A, Mahlaoui N, Hazelaar S, Bakunina K, Slatter M, Collin M, Lum SH, Bigley V, Hauck F, Klein C, Burns SO, Carpenter B, Chakraverty R, Speckmann C, Warnatz K, Cheminant M, Neven B, Beier R, Lange A, Laberko A, Balashov D, Schulz A, Wynn R, Lankester A, Morris EC, March 2021, unpublished data), suggesting that older age per se is not a barrier to HSCT in the absence of IEI-related comorbidities. The challenge remains to identify patients for HSCT as soon as possible after diagnosis or the development of an IEI-related complication that predicts for poor outcome with conservative therapy alone. The immune deficiency and dysregulation activity score before HSCT is currently being evaluated as a tool to predict outcome after HSCT in IEI patients17 (Fox TA, Burns SO, Lever C, Laurence A, Chakraverty R, Grace S, Oliviero F, Workman S, Symes A, Lowe DM, Hough R, Carpenter B, and Morris EC, March 2021, unpublished data).
How we perform HSCT in adult IEI patients
As for all HSCT, donor selection, stem cell source, conditioning regimen, and GVHD prophylaxis all influence outcome and transplant-specific complications. Additional factors may influence donor and graft choice, regimen, and GVHD prophylaxis such as the presence of inherited disease within the family, inflammatory manifestations that may increase GVHD risk, and active infection before HSCT.
Donor selection
Preferred stem cell donors are 10/10 HLA-matched, cytomegalovirus sero-matched, unaffected donors to minimize transplant related mortality (TRM), and GVHD risk while optimizing prompt engraftment and immune reconstitution.46-48 HLA-matched family members should be genetically screened if a known pathogenic variant has been identified in the recipient, and the use of female carriers of X-linked disease as stem cell donors should be avoided where possible. It is notable that matched unrelated donor (MUD) searches for IEI patients are more likely to identify either no or very poor donor options, in part related to patient ethnicity. As related donor options are reduced because of the presence of inherited disease, the use of haploidentical donors and/or mutation carriers may sometimes be the best family option available.49
In general, the use of unrelated donors is common for IEI HSCT, with acceptably good results.10,12,50,51 The use of haploidentical donors is widespread in pediatrics50-52 with excellent outcomes. Despite the growing use of haploidentical donors for adults with IEI, there are currently insufficient published data in older IEI patients to definitively support the use of haploidentical donors in preference to 1 antigen mismatched unrelated donors (1 Ag MMUDs) in adults.
Conditioning regimen selection
IEI patients surviving to adulthood typically have residual functional cellular immunity necessitating conditioning to permit engraftment of allogeneic stem cells and prevent graft rejection. A wide variety of regimens are used for IEI patients with varying degrees of myeloablation from minimally ablative fludarabine with low-dose cylcophosphamide (20-40 mg/kg) used in patients with DNA repair or radio-sensitivity disorders to fludarabine with melphalan (140 mg/m2), treosulfan (30-42 g/m2), or busulfan (area under curve [AUC] 60-70 mg⋅h/L) through to more myeloablative fludarabine with busulfan (AUC 85-95 mg⋅h/L), or fludarabine with treosulfan (30-42 g/m2) and thiotepa (8-10 mg/kg) indicated for younger patients with no preexisting organ damage, where complete donor chimerism is desired for optimal disease correction. We recommend using reduced toxicity or reduced intensity conditioning for the majority of adult IEI patients to limit TRM. Most experience to date has been with fludarabine-based regimens in combination with the alkylating agents busulfan, melphalan, or treosulfan and incorporating serotherapy (alemtuzumab or anti-thymocyte globulin [ATG]/anti-T lymphocyte globulin [ATLG]) for in vivo T-cell depletion as GVHD prophylaxis. Excellent results have also been achieved recently using a radiation-free, serotherapy-free reduced-intensity T cell–replete regimen incorporating pentostatin, low-dose cyclophosphamide, and busulfan with posttransplant cyclophosphamide as GVHD prophylaxis.51 At present, there is insufficient evidence in adult IEI to definitively recommend 1 reduced toxicity regimen over another.
Conditioning regimens using reduced doses of cytoreductive agents are better tolerated in older recipients with higher comorbidities and hence the recommendation, but they carry the risk of failing to eradicate host hematopoiesis.53,54 This can result in mixed chimerism in any lineage, with the T-cell compartment frequently affected unless donor T cells have a significant survival advantage. The impact of mixed chimerism after HSCT varies with underlying IEI and may be deleterious as in GATA2 deficiency, where the risk of myeloid malignancies persists if full donor myeloid chimerism is not achieved. As such, more myeloablative regimens may be indicated in specific patients with the aim of maximizing engraftment and full lineage donor chimerism.
As a general principle, high degrees of donor chimerism are more likely to result in sustained functional correction of the underlying IEI and optimal long-term outcome as has been documented in patients with WAS;32,44,55 however, there may be some IEIs for which this is not true. There is good evidence in X-CGD that the use of carriers as donors may not result in full immunologic correction, even in the presence of full donor chimerism (depending on carrier neutrophil function tests). However, in other X-linked and autosomal recessive disorders, carriers may be used as donors if the relevant functional immunologic assays are normal. It is not yet known what degree of chimerism is sufficient to correct the clinical phenotype in gain of function (GOF) diseases.
Serotherapy and GVHD prophylaxis
Serotherapy with ATG/Thymoglobuline (polyclonal rabbit antithymocyte globulin), ATLG/Grafalon (anti–T-lymphocyte globulin), or alemtuzumab (CD52 monoclonal antibody) is commonly used to prevent both GVHD and graft rejection, being of most value in unrelated and mismatched related donor transplants. Serotherapy may also be useful in IEI with inflammatory manifestations. These biologics have different properties, and exposure following standard dosing is highly variable, with studies demonstrating both patient weight and lymphocyte count before infusion as influential.56-58 Timing of administration is also important, particularly when unmanipulated grafts are infused and the concentration of ATG/ATLG or alemtuzumab on day 0 influences both engraftment and prevention of GVHD through donor T-cell depletion. Overexposure can lead to prolonged lymphocyte depletion, delayed immune reconstitution, and mixed chimerism. Personalized dosing using real-time pharmacokinetic and pharmacodynamic analysis is not widely available.
If no serotherapy is used in unrelated, mismatched related, or haploidentical HSCT, additional measures are required to prevent GVHD. Strategies such as posttransplant cyclophosphamide and alpha/beta T cell receptor depletion to selectively deplete donor-derived alloreactive T cells have facilitated the use of alternative donors in older recipients with hematologic malignancies, without prohibitive risks of GVHD and/or graft rejection,59 and in pediatric series, excellent results have been achieved in IEI.50,52,60,61 However, at the time of writing, there remains little cumulative experience transplanting older adults with IEIs using haploidentical donors, and we use these donors in younger adults where no other options are available. Further studies are warranted and may result in the wider adoption of haplo-HSCT for older adults with IEIs in the future.
Specific HSCT management for adults with IEI to optimize outcome
Identification of IEI-specific comorbidities
In addition to routine pre-HSCT investigations, we recommend documenting the presence or absence of specific pathogens, antimicrobial sensitivities, and/or degree of organ damage in adult IEI patients. Specialist infectious disease advice should be sought for patients with a history of refractory or atypical infections preceding transplant. Table 1 lists some common noninfectious comorbidities seen in adult IEI patients, which increase HSCT risk and if present will alter the risk:benefit ratio and may influence the final transplant decision and/or conditioning regimen selection. Figure 1 illustrates the importance of pre-HSCT multidisciplinary working to optimise patient management, and its potential impact on the decision to proceed. For patients proceeding to HSCT with preexisting end-organ damage, the consent process must involve a clear discussion about which disease-associated symptoms/complications can be improved by transplant and which cannot (eg, bronchiectasis/pulmonary fibrosis, gut strictures). The same discussion is required for extrahematologic complications of IEI.
Common noninfectious comorbidities to consider in adults with IEIs that confer worse outcome for HSCT
| System | Complications commonly seen in adults with IEI | Investigations recommended | Influence on transplant planning |
|---|---|---|---|
| Pulmonary | Structural lung disease including: • Bronchiectasis • Fibrosis • Pneumatocele | Lung function: spirometry and gas transfer. High resolution chest CT. Bronchoscopy and BAL if suspect active undiagnosed infection. | FVC and FEV1 < 60% predicted: acceptable if not oxygen-dependent and possibility for stabilization post-HSCT. Ensure optimal management of bronchiectasis and/or ILD before HSCT. Sputum surveillance to inform peritransplant antimicrobial prophylaxis and treatment. |
| Inflammatory lung disease including: • Granulomatous interstitial lung disease • Cryptogenic organizing pneumonia | Lung function: gas transfer is a good marker of extent of ILD. High resolution CT chest. Bronchoscopy and BAL if suspect active undiagnosed infection. | DLCO < 50% predicted: acceptable if not oxygen-dependent and possibility for stabilization post-HSCT. ILD anticipated to improve with immunosuppression conferred by conditioning. Associated structural change, such as fibrosis, may be a barrier to HSCT (see above). Occasionally thoracic surgery indicated before HSCT (eg, resection of infected cavity/lobe). | |
| Renal | Chronic renal impairment including: • Autoimmune renal disease • Vasculitis • Drug induced injury | Urea, creatinine, electrolytes and estimated GFR. Renal biopsy (occasionally indicated when renal diagnosis unclear). | GFR < 30 mL/kg/min poses a significant barrier to HSCT because of reduced drug tolerance and increased risk of sepsis associated acute kidney injury (AKI). For patients with GFR < 30 mL/kg/min and no other severe end organ damage, peritransplant hemofiltration can be considered, but requires highly specialist care and complicates drug dosing especially for chemotherapy. |
| Hepatic | Chronic liver disease including: • Nodular regenerative hyperplasia • Fibrosis/cirrhosis • Portal hypertension ± ascites | Liver function testing to include transaminases, alkaline phosphatase, bilirubin, albumin, INR, Hepatitis B and C DNA/RNA* Liver ultrasound. Fibroscan measuring liver stiffness. Liver biopsy. Portal venous pressure measurements. If portal hypertension, upper endoscopy for esophageal varices. | Severe chronic liver disease is an absolute barrier to HSCT including: • Liver synthetic failure • Decompensated portal hypertension with ascites • Severe portal hypertension with large splenomegaly • Cirrhosis Decision making requires involvement of an experienced hepatology team. Prior liver transplant or surgical TIPPS procedure (for portal hypertension) can be considered but there is little data for these procedures in adults with IEI to date, making this high risk. |
| Active inflammation • Autoimmune hepatitis | As above Also tissue specific autoantibodies. | Control of active inflammation using immunosuppression advised prior to HSCT. Decision making requires involvement of an experienced hepatology team and prior liver transplant may be required for refractory inflammation leading to liver failure. | |
| Gastrointestinal | Chronic diarrhea Malabsorption and poor nutrition | Stool samples for microscopy and culture/polymerase chain reaction to exclude bacterial and parasitic pathogens including disease specific considerations: eg, cryptosporidium in CD40L deficiency. Polymerase chain reaction for viruses, including norovirus. Vitamin levels. | Infectious disease or microbiology input indicated for chronic or relapsing infections. Eradication pre-HSCT may not be feasible but peri- and post-HSCT prophylaxis may be required. Nutrition should be optimized if possible, prior to HSCT, including use of parenteral nutrition if required. |
| Active inflammation/colitis • Inflammatory bowel disease • Granulomatous inflammation | Fecal calprotectin Upper and lower endoscopy with biopsies if pathogens excluded and diagnosis unclear. | Conditions with active inflammation typically improve with immunosuppression conferred by conditioning. | |
| Spleen | Splenomegaly for example • As a feature of the specific IEI • With autoimmune cytopenias • Secondary to liver disease and portal hypertension (see above) | Ultrasound or computed tomography scans of abdomen to ascertain size. Liver investigations if liver disease suspected (see above) | Large splenomegaly increases risk of engraftment failure and is associated with poor count recovery. If secondary to liver disease, follow advice for liver complications (see above) If related to immune dysregulation of the underlying IEI, consider whether size can be reduced prior to HSCT eg with immunosuppression such as sirolimus or rituximab or with control of autoimmune cytopenias. Splenectomy not advised due to risk of post-HSCT infections, in particular pneumococcal sepsis. Embolization also not advised due to risk of abscess formation, unless urgent need to reduce spleen size for control of severe refractory autoimmune cytopenia. |
| Prior splenectomy | None | Ensure pneumococcal antibiotic prophylaxis post HSCT, typically lifelong. Ensure pneumococcal vaccination post-HSCT with confirmation of protective antibody response. |
| System | Complications commonly seen in adults with IEI | Investigations recommended | Influence on transplant planning |
|---|---|---|---|
| Pulmonary | Structural lung disease including: • Bronchiectasis • Fibrosis • Pneumatocele | Lung function: spirometry and gas transfer. High resolution chest CT. Bronchoscopy and BAL if suspect active undiagnosed infection. | FVC and FEV1 < 60% predicted: acceptable if not oxygen-dependent and possibility for stabilization post-HSCT. Ensure optimal management of bronchiectasis and/or ILD before HSCT. Sputum surveillance to inform peritransplant antimicrobial prophylaxis and treatment. |
| Inflammatory lung disease including: • Granulomatous interstitial lung disease • Cryptogenic organizing pneumonia | Lung function: gas transfer is a good marker of extent of ILD. High resolution CT chest. Bronchoscopy and BAL if suspect active undiagnosed infection. | DLCO < 50% predicted: acceptable if not oxygen-dependent and possibility for stabilization post-HSCT. ILD anticipated to improve with immunosuppression conferred by conditioning. Associated structural change, such as fibrosis, may be a barrier to HSCT (see above). Occasionally thoracic surgery indicated before HSCT (eg, resection of infected cavity/lobe). | |
| Renal | Chronic renal impairment including: • Autoimmune renal disease • Vasculitis • Drug induced injury | Urea, creatinine, electrolytes and estimated GFR. Renal biopsy (occasionally indicated when renal diagnosis unclear). | GFR < 30 mL/kg/min poses a significant barrier to HSCT because of reduced drug tolerance and increased risk of sepsis associated acute kidney injury (AKI). For patients with GFR < 30 mL/kg/min and no other severe end organ damage, peritransplant hemofiltration can be considered, but requires highly specialist care and complicates drug dosing especially for chemotherapy. |
| Hepatic | Chronic liver disease including: • Nodular regenerative hyperplasia • Fibrosis/cirrhosis • Portal hypertension ± ascites | Liver function testing to include transaminases, alkaline phosphatase, bilirubin, albumin, INR, Hepatitis B and C DNA/RNA* Liver ultrasound. Fibroscan measuring liver stiffness. Liver biopsy. Portal venous pressure measurements. If portal hypertension, upper endoscopy for esophageal varices. | Severe chronic liver disease is an absolute barrier to HSCT including: • Liver synthetic failure • Decompensated portal hypertension with ascites • Severe portal hypertension with large splenomegaly • Cirrhosis Decision making requires involvement of an experienced hepatology team. Prior liver transplant or surgical TIPPS procedure (for portal hypertension) can be considered but there is little data for these procedures in adults with IEI to date, making this high risk. |
| Active inflammation • Autoimmune hepatitis | As above Also tissue specific autoantibodies. | Control of active inflammation using immunosuppression advised prior to HSCT. Decision making requires involvement of an experienced hepatology team and prior liver transplant may be required for refractory inflammation leading to liver failure. | |
| Gastrointestinal | Chronic diarrhea Malabsorption and poor nutrition | Stool samples for microscopy and culture/polymerase chain reaction to exclude bacterial and parasitic pathogens including disease specific considerations: eg, cryptosporidium in CD40L deficiency. Polymerase chain reaction for viruses, including norovirus. Vitamin levels. | Infectious disease or microbiology input indicated for chronic or relapsing infections. Eradication pre-HSCT may not be feasible but peri- and post-HSCT prophylaxis may be required. Nutrition should be optimized if possible, prior to HSCT, including use of parenteral nutrition if required. |
| Active inflammation/colitis • Inflammatory bowel disease • Granulomatous inflammation | Fecal calprotectin Upper and lower endoscopy with biopsies if pathogens excluded and diagnosis unclear. | Conditions with active inflammation typically improve with immunosuppression conferred by conditioning. | |
| Spleen | Splenomegaly for example • As a feature of the specific IEI • With autoimmune cytopenias • Secondary to liver disease and portal hypertension (see above) | Ultrasound or computed tomography scans of abdomen to ascertain size. Liver investigations if liver disease suspected (see above) | Large splenomegaly increases risk of engraftment failure and is associated with poor count recovery. If secondary to liver disease, follow advice for liver complications (see above) If related to immune dysregulation of the underlying IEI, consider whether size can be reduced prior to HSCT eg with immunosuppression such as sirolimus or rituximab or with control of autoimmune cytopenias. Splenectomy not advised due to risk of post-HSCT infections, in particular pneumococcal sepsis. Embolization also not advised due to risk of abscess formation, unless urgent need to reduce spleen size for control of severe refractory autoimmune cytopenia. |
| Prior splenectomy | None | Ensure pneumococcal antibiotic prophylaxis post HSCT, typically lifelong. Ensure pneumococcal vaccination post-HSCT with confirmation of protective antibody response. |
BAL, broncho-alveolar lavage; CT, computerised tomography; DLCO, diffusion capacity of lung for carbon monoxide; FVC, forced vital capacity; GFR, glomerular filtration rate; ILD, intersitial lung disease; INR, international normalised ratio; TIPPS, transjugular intrahepatic portosystemic shunt.
Serology is unreliable in IEIs because antibody production is commonly impaired or patients are on immunoglobulin replacement therapy. Autoantibodies are usually unhelpful for the same reasons.
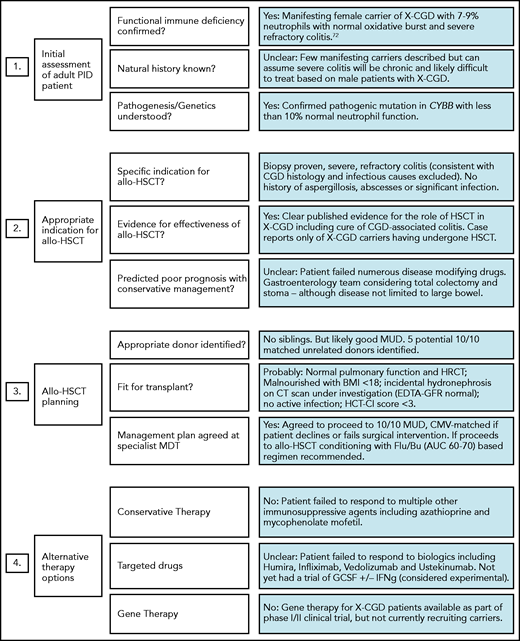
Example of adult IEI HSCT MDT impact on patient management. MDT aims include (i) standardizing practice; (ii) sharing experience in the clinical management of rare HSCT indications; and (iii) providing patient specific advice regarding the risk of transplant, choice of conditioning regimens, and selection of most appropriate transplant center. Green, amber, and red boxes represent a traffic light system, with green being favorable and red unfavorable.
Example of adult IEI HSCT MDT impact on patient management. MDT aims include (i) standardizing practice; (ii) sharing experience in the clinical management of rare HSCT indications; and (iii) providing patient specific advice regarding the risk of transplant, choice of conditioning regimens, and selection of most appropriate transplant center. Green, amber, and red boxes represent a traffic light system, with green being favorable and red unfavorable.
Optimization of IEI-related complications before HSCT
Where possible, control of infection, autoimmunity, and/or inflammation should be achieved before transplant. Reducing the inflammatory cytokine milieu at the time of transfer of allogeneic cells reduces the risk of acute GVHD and ensures end organ function is optimized before transplant. Patients with granulomatous and lymphoproliferative manifestations of IEIs, including granulomatous lymphocytic inflammatory lung disease, should receive immunosuppressive therapies to reduce inflammation and optimize end organ function before HSCT. Preexisting IEI-associated malignancies should be treated and in remission as per routine practice in HSCT for lymphoid malignancies. However, in some cases, IEI-associated malignancies are challenging to treat as cytopenias, or concurrent infection may preclude optimal antineoplastic therapy because of excess toxicity. Such patients are more likely to proceed to HSCT without achieving a complete remission. When considering lymphoma, as with non–IEI-associated lymphomas, the risk of relapse after HSCT is lower for patients who have chemo-responsive disease and reduced disease burden before HSCT. Patients with IEI-associated HLH should be in remission or have stably controlled disease before HSCT because the outcome for patients transplanted with frank HLH is poor.62,63 For patients with Epstein-Barr virus (EBV) handling disorders, the inclusion of rituximab in the conditioning regimen can bridge the gap until functional immune reconstitution is achieved after transplant.
IEI-specific peri-HSCT management
In addition to routine HSCT management, regular posttransplant monitoring for recurrence of previous, persistent, or latent opportunistic pathogens is indicated on a per-patient basis. We recommend the continued use of prophylactic antimicrobials until cessation of systemic immune suppression as either GVHD prophylaxis or treatment and the continuation of IRT until a minimum of 6 months after HSCT.
Post-HSCT monitoring and long-term follow-up
Standard post-HSCT monitoring
Patients require standard post-HSCT follow-up and monitoring to evaluate for HSCT-related complications such as GVHD, reactivation of latent viruses, late graft failure, secondary malignancies, and other known late effects.
IEI-specific post-HSCT monitoring
Other important end points for IEI patients include the reconstitution of normal pathogen-specific immunity, adequate B-cell reconstitution and immunoglobulin production, resolution of autoimmunity and/or inflammation, and reduction in future malignancy risk. For patients with EBV handling disorders (such as CD27 or CD70 deficiency, XLP1 and XIAP deficiency), regular monitoring for EBV reactivation is essential because persistent EBV viremia is associated with HLH and lymphoproliferation.64,65 Other additional monitoring is disease specific and influenced by the degree of lineage-specific chimerism achieved after HSCT. For example, patients transplanted for GATA2 deficiency who do not achieve full myeloid chimerism after HSCT will be at risk of relapse of MDS or acute myeloid leukemia and should be monitored appropriately.
In adult IEIs, the indication for transplant is often the prevention of progressive decline in quality of life secondary to the accumulation of IEI-related medical complications. Because of this, we recommend including patient-reported outcomes and quality-of-life assessments when evaluating success.66
Chimerism monitoring
During the first year after reduced-intensity HSCT, chimerism often fluctuates. The use of donor lymphocyte infusions to promote conversion from mixed chimerism to full donor chimerism carries a risk of GVHD and typically is only used when worsening mixed chimerism raises concerns of incipient graft rejection. However, recently emerging data from the long-term follow-up of pediatric HSCT recipients indicate that persistent mixed chimerism after transplant is associated with reduced event-free survival after as long as 20 years (Day J, Elfeky R, Nicholson B, Goodman R, Pearce R, Fox TA, Worth A, Booth C, Veys P, Carpenter B, Hough R, Gaspar B, Titman P, Ridout D, Workman S, Hernandes F, Sandford K, Laurence A, Campbell M, Burns SO, Morris EC, September 2020, unpublished data) and late complications such as autoimmunity, which has been observed in patients transplanted for WAS but not for other IEIs.44,45,65 For patients with phagocytic defects, achieving high level stable myeloid chimerism is essential. Studies in female carriers of X-linked CGD have identified inflammatory disease similar to that seen in male patients in carriers with extreme degrees of lyonization (resulting in <20% to 30% normally functioning phagocytes), although the risk of serious infection is rarely present if >10% of circulating neutrophils are functional.67-69 Carriers of X-linked diseases typically have 50% normal and 50% abnormal cells in the relevant lineage, whereas in autosomal recessive disorders carriers typically have normal function in 100% of cells. Therefore, the impact of post-HSCT mixed chimerism will depend on the specific IEI if a carrier is used as stem cell donor. For rarer IEIs such as activated phosphoinositide 3-kinase delta syndrome, cytotoxic T lymphocyte-associated protein-4 deficiency, lipopolysaccharide responsive and beige-like anchor protein deficiency, dedicator of cytokinesis 8 deficiency, and signal transducer and activator of transcription 1 GOF, there are insufficient data regarding the optimal lineage-specific chimerism required for long-term immunologic correction and cure, although insight into disease-specific pathophysiology may predict which lineages are critical to correct. For example, even small populations of residual recipient cells may be problematic in GOF diseases.
Reconstitution of normal immune function
Lineage specific donor chimerism can be used as a surrogate marker for functional correction, but proof of transplant efficacy relies on immunologic assessment (eg, neutrophil function tests, response to vaccination, T-cell proliferation, normalization of lymphocyte subsets, and assessment of thymopoiesis) and clinical responses. Post-HSCT vaccination schedule is as for other HSCT indications. Vaccination should not be undertaken until patients are off IRT, typically 6 to 12 months after HSCT. In specific cases, additional vaccination may be warranted, for example, HPV vaccination in patients with IEIs conferring HPV susceptibility such as GATA2 deficiency. Protective specific antibody responses to vaccination should be documented where tests are available, for example, serotype-specific pneumococcal antibodies. Currently, specific vaccination of donors to provide pathogen-specific immunity is not routine.
Malignancy
Patients with previous HPV-associated dysplasia or malignancy should continue to have regular surveillance (eg, cervical smears, colposcopy ± biopsy). Patients with previous lymphoma should have regular imaging (computed tomography/positron emission tomography or positron emission tomography/magnetic resonance imaging) as per standard post-HSCT practice.
Specific HSCT management of our cases
With suitably matched donors, we expect TRM to be in the region of 10% to 15% based on our own center’s HSCT outcome data for carefully selected adult patients in the absence of major end organ dysfunction or active HLH.12
Case 1 (42 years, WAS) has moderately severe bronchiectasis but preserved pulmonary function with an forced expiratory volume (FEV1) > 50% predicted. With a well-matched unrelated donor, we would recommend HSCT using a reduced toxicity conditioning regimen with serotherapy or posttransplant cyclophosphamide as GVHD prophylaxis to prevent disease progression and predict a TRM of 15% to 20% in view of the bronchiectasis.
Case 2 (24 years, CGD) had excellent end organ function and proceeded to an unrelated donor HSCT to restore pathogen immunity and achieve remission of his refractory CGD colitis. Most CGD patients transplanted to date have received fludarabine and targeted busulfan (AUC 70) with either ATG (for MRD) or alemtuzumab (for MUD/MMUD).10,12 He is now 19 months after HSCT with tri-lineage full donor chimerism, no active colitis, off immune suppression, and no further infections. He is exercising again and well enough to apply for work.
Case 3 (26 years, GATA2 deficiency) had a number of moderate comorbidities when referred for HSCT. She underwent 1Ag MMUD with fludarabine/melphalan-alemtuzumab conditioning. No additional rituximab was given before transplant in the absence of EBV viremia. She is now 7 years after HSCT, off immune suppression, and with no GVHD. Complete resolution of HPV disease occurred at 5 years after HSCT, and she has since had 2 normal cervical smears. She is currently deciding whether to proceed to in vitro fertilisation with a limited number of cryopreserved eggs with or without preimplantation genetic diagnosis.
Finally, case 4 (35 years, complex CVID) represents a major challenge. Although the published HSCT data for CVID patients included a large number of patients who received myeloablative transplants, the outcomes were significantly poorer than for other IEIs,14 and prospective studies are urgently required to assess safety and efficacy of more modern regimens in carefully selected patients. Our patient has very significant comorbidities (bronchiectasis, chronic norovirus infection, portal hypertension, massive splenomegaly, ascites, and noncirrhotic liver fibrosis), which predict an excessive TRM even with reduced intensity conditioning. However, because of preserved synthetic liver function, he does not currently meet the criteria for liver transplantation. His best stem cell donor is a 1 Ag MMUD, and we would not recommend proceeding to HSCT in this case without prior liver transplant.
When we consider gene therapy as an alternative to HSCT
Some patients referred to us have a clear indication for HSCT but no well-matched donor. In these cases, the decision between using multiple mismatched cord units, a mismatched unrelated donor, a haploidentical donor, or to consider gene therapy (GT) is complex. Currently, GT approaches where autologous hematopoietic stem cells (HSCs) are genetically modified ex vivo using viral vectors encoding a wild-type version of the mutated gene are only available for a few monogenic diseases including X-linked severe combined immune deficiency, adenosine deaminase deficiency severe combined immune deficiency, WAS, and X-CGD. Only 1 GT is currently licensed (Strimvelis for adenosine deaminase deficiency severe combined immune deficiency), although at the time of writing, it is unavailable while an occurrence of insertional mutagenesis is being investigated, and for other diseases, patients will need to be recruited into clinical trials. The potential advantage of GT is the requirement for less immunosuppressive conditioning and no risk of GVHD because it uses autologous HSCs. However, engraftment of gene-modified HSCs requires myeloablative preconditioning chemotherapy that is associated with significant toxicities in older patients. Furthermore, current GT approaches result in only partial correction of autologous cells generating a setting equivalent to achieving mixed chimerism after HSCT. The degree of correction is influenced by ex vivo HSC transduction efficiency, degree of engraftment of the modified HSCs, potential survival advantage (or not) of the gene-corrected immune cells, and durability of gene expression. The impact of these factors on clinical outcome after GT is likely to vary among IEIs. GT has been successfully used in adults for WAS70,71 and X-linked CGD,72 where appropriately matched allogeneic stem cell donors were not available. This is an appealing option for adults with significant comorbidities and/or poorly matched donors, and if long-term correction and immune reconstitution is proven to be effective and durable, GT may become the preferred option for adults with IEIs in the future.
Conclusion
The application of HSCT to adults with IEIs is a rapidly emerging field. Decision making is complex given the heterogenous nature of IEI, the variety of associated complications, and the paucity of outcome data. Despite this, our experience is that HSCT can be delivered as successfully to carefully selected adults as to children with IEI. Identifying adult IEI patients for whom HSCT is appropriate requires both detailed understanding of the disease and the transplant process and should involve the combined input of immunologists and transplant physicians, ideally in a joint clinical setting. Over the coming years, we expect that HSCT for adult IEI will expand both in terms of patient numbers and range of conditions treated. We also expect gene therapy and gene editing approaches to become more widely available as alternative options. Although there remains a pressing need to enrich outcome data both for conservative management and corrective therapies to help our patients make the most informed choice for their care, these are promising times for adults with IEI.
Authorship
Contribution: E.C.M. and S.O.B. co-wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Emma C. Morris, Department of Immunology, UCL Institute of Immunity & Transplantation, 2nd Floor, Royal Free Hospital, Rowland Hill St, London NW3 2PF, United Kingdom; e-mail: e.morris@ucl.ac.uk.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal