

Key Points
The platelet αIIbβ3 integrin receptor is produced in various covalent forms that have different functions.
One-third of αIIbβ3 molecules are missing an αIIb disulfide bond that changes distribution and function of the receptor.

Abstract
The αIIbβ3 integrin receptor coordinates platelet adhesion, activation, and mechanosensing in thrombosis and hemostasis. Using differential cysteine alkylation and mass spectrometry, we have identified a disulfide bond in the αIIb subunit linking cysteines 490 and 545 that is missing in ∼1 in 3 integrin molecules on the resting and activated human platelet surface. This alternate covalent form of αIIbβ3 is predetermined as it is also produced by human megakaryoblasts and baby hamster kidney fibroblasts transfected with recombinant integrin. From coimmunoprecipitation experiments, the alternate form selectively partitions into focal adhesions on the activated platelet surface. Its function was evaluated in baby hamster kidney fibroblast cells expressing a mutant integrin with an ablated C490-C545 disulfide bond. The disulfide mutant integrin has functional outside-in signaling but extended residency time in focal adhesions due to a reduced rate of clathrin-mediated integrin internalization and recycling, which is associated with enhanced affinity of the αIIb subunit for clathrin adaptor protein 2. Molecular dynamics simulations indicate that the alternate covalent form of αIIb requires higher forces to transition from bent to open conformational states that is in accordance with reduced affinity for fibrinogen and activation by manganese ions. These findings indicate that the αIIbβ3 integrin receptor is produced in various covalent forms that have different cell surface distribution and function. The C490, C545 cysteine pair is conserved across all 18 integrin α subunits, and the disulfide bond in the αV and α2 subunits in cultured cells is similarly missing, suggesting that the alternate integrin form and function are also conserved.
Introduction
Hemostasis is a complex interplay of the vascular endothelium, platelets, and coagulation factors to arrest bleeding. This process may lead to thrombosis and obstruction of coronary or cerebral blood vessels, the precipitating event in myocardial infarction and ischemic stroke that accounts for ∼1 in 4 of all deaths each year. Individuals with a heightened tendency to form thrombi also have an accelerated development of atherosclerosis and are at increased risk of acute coronary syndrome and myocardial infarction. Blood platelets, anucleate cells derived from bone marrow megakaryocytes, are the effector cells of thrombosis.
Resting platelets circulate as spherical biconcave-shaped cells in the vasculature with a life span of 8 to 10 days. They are captured from the blood at a site of vascular injury by von Willebrand factor, leading to release of agonists such as thromboxane A2 and adenosine 5′-diphosphate that stimulate platelet G protein–coupled and purinergic receptors, resulting in platelet activation and change in shape. Activated platelets form a plug at the site of vessel injury through cross-linking of platelet αIIbβ3 integrin by blood fibrinogen. Platelets contain 5 different integrins, and αIIbβ3 is by far the most abundant, with each platelet expressing 80 000 to 100 000 molecules. This integrin is critical for thrombus formation and is the target of antithrombotic agents in clinical use for acute coronary syndrome.
αIIbβ3 integrin has a remarkable form–function relationship that has been studied extensively. The receptor undergoes several conformational transitions that have distinct ligand affinities, signaling functions, and force-sensing capacities.1-3 The conformational transitions in αIIbβ3 integrin have been described in terms of changes in noncovalent interactions such as breaking and forming of hydrogen, ionic, and hydrophobic bonds and van der Waals interactions. Recently, changes in a covalent bond have been described. The secreted platelet oxidoreductase, ERp5, mediates release of fibrinogen from activated αIIbβ3 integrin through cleavage of the β3 subunit C177-C184 disulfide bond.4 In this study, we have identified an alternate integrin form in platelets that is defined by a missing disulfide bond in the αIIb subunit.
The αIIb subunit consists of a β-propeller headpiece followed by Thigh, Genu, and 2 Calf domains. Extension of the subunit involves unbending at the Genu or knee joint. We quantified the redox state of 8 of the 9 disulfide bonds in the αIIb subunit of healthy human donor platelet αIIbβ3 integrin and found that the C490-C545 disulfide bond in the Thigh domain is missing in ∼1 in 3 molecules of the total integrin pool. This alternate covalent form of αIIbβ3 is predetermined and selectively partitions into focal adhesions on the activated platelet surface, where it has extended residency time. The alternate form also has different conformational dynamics that correlate with reduced fibrinogen binding and outside-in signaling. These findings indicate that the αIIbβ3 integrin receptor is produced in various covalent forms that have different functions.
Methods
Blood collection and processing
Blood was obtained from healthy volunteers with the approval of The University of Sydney Human Research Ethics Committee (protocol no. X17-0390). All procedures were in accordance with the 1983 Declaration of Helsinki. Whole blood was drawn into citrate tubes and platelet-rich plasma collected by centrifugation at 200g for 20 minutes at 22°C. Prostaglandin E1 (1 μM) was added and incubated for 30 minutes at 37°C before platelets were collected by centrifugation at 800g for 15 minutes and resuspended in prewarmed N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)-buffered Tyrode’s solution containing 5 mM glucose (0.5 mL per 5 mL of blood). When indicated, platelets were activated by addition of 1 U/mL thrombin, 5 µg/mL equine tendon collagen, type I (HORM, Takeda), 10 mM MgCl2, and 10 mM CaCl2, without or with 1 mM Gly-Pro-Arg-Pro peptide (MilliporeSigma) for 6 minutes. Thrombin was inactivated after 6 minutes with 50 µM PPACK (Sapphire Bioscience).
Quantification of the redox states of αIIb disulfide bonds
Washed platelets from 5 mL of blood or 107 MEG-01, U251, HUVEC/TERT2, or baby hamster kidney-21 (BHK) cells were labeled with 2-iodo-N-phenylacetamide (IPA, 4 mM) for 1 hour at 22°C and collected by centrifugation at 1500g for 10 minutes. Cells were lysed with 0.2 mL of 20 mM HEPES pH 7.4 buffer containing 2% NP40, 150 mM NaCl, 2 mM EDTA, 50 mM N-ethylmaleimide and proteinase inhibitor cocktail (cOmplete Mini EDTA-Free Protease Inhibitor Cocktail, PhosStop Phosphatase Inhibitor from Roche and 1 mM phenylmethylsulfonyl fluoride), and lysate collected by centrifugation at 10000g for 20 minutes at 4°C. Lysate and anti-β3 (AP3, RRID:AB_2056630), anti-α2β1 (clone P1E6; Abcam ab24697), or antitalin (clone 8d4, ab157808; Abcam) antibody (10 µg per sample) were mixed in 0.5 mL of wash buffer (25 mM Tris-HCl pH 7.4 buffer containing 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 5% glycerol) and rotated overnight at 4°C. The αIIbβ3, αVβ3, or α2β1 integrin was collected on 80 μL of protein A/G agarose with rotation for 2 hours at 25°C. The beads were washed 3 times with wash buffer and 3 times with phosphate-buffered saline (PBS). 12C-IPA (4 mM) was added to the beads and incubated with rotation for 1 hour at 22°C. The integrin and antibody were released from the beads by boiling in 40 µL 2 × NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Thermo Fisher Scientific) for 20 minutes at 70°C and the integrin resolved on sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE). In some instances, membrane surface integrins were isolated from platelets by labeling washed platelets (isolated as described earlier) with 200 µM cell impermeable Sulfo-NHS-Biotin (Thermo Fisher Scientific) for 15 minutes at 4°C. The reaction was quenched and unlabeled biotin removed by washing twice in PBS containing 100 mM glycine. Biotin-labeled surface integrins were immunoprecipitated by using M-280 streptavidin Dynabeads (Thermo Fisher Scientific) according to manufacturer’s instructions.
The αIIb, αV, or α2 bands on SDS-PAGE were excised, washed, and dried in 100% acetonitrile. All band washings were done 3 times with 25 mM sodium bicarbonate buffer containing 50% acetonitrile while being shaken at 1400 rpm for 10 minutes. All reactions were performed in the dark. To measure S-nitrosation of αIIbβ3, dried gel pieces were incubated with 20 mM sodium ascorbate for 1 hour at 25°C, washed, dried in 100% acetonitrile, and the resulting free thiols alkylated with 55 mM iodoacetamide for 1 hour at 25°C. Washed and dried gel pieces were incubated with 40 mM dithiothreitol for 30 minutes at 56°C and washed again. The fully reduced protein was alkylated for 1 hour at 22°C with 5 mM IPA in which all 6 carbon atoms of the phenyl ring have a mass of 13 (13C-IPA; Cambridge Isotopes). The gel slices were washed, dried, and deglycosylated overnight using 5 units of PNGase F (MilliporeSigma) at 37 °C, before digestion of the integrin with 12.5 ng/mL of chymotrypsin (Roche) in 25 mM NH4CO2 and 10 mM CaCl2 for 5 hours at 37°C, followed by digestion with 12.5 ng/mL of trypsin overnight at 25°C. Reactions were stopped by adding 5% (v/v) formic acid and peptides eluted from the gel slices with 5% formic acid and 50% (v/v) acetonitrile. Peptides were desalted by using C18 ZipTip (Merck Millipore). Liquid chromatography, mass spectrometry, and data analysis were performed as described.5 Briefly, 200 ng of peptides were resolved on a 35 cm × 75 μm C18 reverse-phase analytical column using a 2% to 35% acetonitrile gradient over 22 minutes with a flow rate of 250 nL/min (Ultimate 3000 HPLC; Thermo Fisher Scientific). The peptides were ionized by electrospray ionization at +2.0 kV. Tandem mass spectrometry (MS/MS) analysis was performed on a Q Exactive Plus mass spectrometer using higher energy collisional induced dissociation fragmentation. The data-dependent acquisition method acquired MS/MS spectra of the top 10 most abundant ions with charged state ≥2 at any one point during the gradient. MS/MS spectra were searched against the SwissProt reference proteome using an external search engine Mascot (version 2.7, Matrix Science) or αIIbβ3 protein sequences (UniProt identifiers P08514 and P05106) using Byonic (version 3.0; Protein Metrics). Precursor mass tolerance and fragment tolerance were set at 10 ppm and the precursor ion charge state to 2+, 3+, and 4+. Variable modifications were defined as oxidized Met, deamidated Asn/Gln, iodoacetanilide derivative Cys, 13C-iodoacetanilide derivative Cys, and carbamidomethyl Cys with full trypsin and chymotrypsin cleavage of up to 3 missed cleavages. The abundance of the different redox forms of the cysteines were calculated from the relative ion abundance of peptides labeled with 12C-IPA and/or 13C-IPA. The criteria for αIIb cysteine containing peptides analyzed to determine disulfide bond redox state are described in supplemental Table 1 (available on the Blood Web site). To calculate ion abundance of peptides, extracted ion chromatograms were generated by using the XCalibur Qual Browser software version 2.1.0 (Thermo Fisher Scientific). The area was calculated by using the automated peak detection function built into the software. The data were searched for peptides containing unlabeled (free) Cys thiols. These were only detected in 2 peptides in trace amounts: peptides NIQM(oxidation)CVGATGH and LICNQK with an incidence of 0.23% and 0.65% of the total peptide species, respectively. This result indicates that alkylation of unpaired Cys residues by 12C-IPA or 13C-IPA was complete in the protein.
αIIb disulfide-linked peptide analysis
Purified human αIIbβ3 (3 µg) was incubated with 5 mM 12C-IPA in PBS containing 10% dimethyl sulfoxide for 1 hour at 25°C in the dark. Protein was denatured in NuPAGE LDS sample buffer for 15 minutes at 70°C and resolved on SDS-PAGE. Gel slices containing αIIb were excised, washed, and dried before deglycosylation overnight using 5 units of PNGase F at 37°C. Samples were digested with 12.5 ng/mL of chymotrypsin in 25 mM NH4CO2 and 10 mM CaCl2 for 5 hours at 37°C, followed by digestion with 12.5 ng/mL of trypsin overnight at 25°C. Reactions were stopped by adding 5% (v/v) formic acid and peptides eluted from the gel slices with 5% formic acid and 50% (v/v) acetonitrile. Peptides were desalted by using C18 ZipTip. Peptides in 0.1% formic acid (final volume, 12 μL) were resolved on a 35 cm × 75 μm C18 reverse-phase analytical column using a 2% to 35% acetonitrile gradient over 22 minutes at a flow rate of 300 nL/min (Ultimate 3000 HPLC), ionized by electrospray ionization at +2.0 kV and analyzed on a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific) using higher energy collisional induced dissociation fragmentation. The data-dependent acquisition method acquired MS/MS spectra of the top 5 most abundant ions with charged state ≥2 at any one point during the gradient. Disulfide-linked peptides were searched against human αIIb sequence using Byonic analysis software. Precursor mass tolerance and fragment tolerance were set at 10 ppm and 0.6 Da, respectively. Variable modifications were defined as oxidized Met, deamidated Asn/Gln, iodoacetanilide derivative Cys, and carbamidomethyl Cys with full trypsin and chymotrypsin cleavage of up to 3 missed cleavages. The false discovery rate was set at 0.01.
Cell lines and culture
MEG-01 megakaryoblasts from American Type Culture Collection were maintained in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco), GlutaMAX (Gibco), and 1% (v/v) penicillin-streptomycin solution (Thermo Fisher Scientific) at 37°C and 5% carbon dioxide (CO2). At passaging, cells were scraped to create a cell suspension and split into a new flask. U-251 cells from American Type Culture Collection were grown in Dulbecco’s modified Eagle medium supplemented with 10% (v/v) FBS, GlutaMAX, and 1% (v/v) penicillin-streptomycin solution at 37°C and 5% CO2. HUVEC/TERT2 cells were a kind gift from Mathias Francois.
Mammalian cell expression of wild-type and C490A and C545A mutant αIIbβ3 integrins
Complementary DNA (cDNA) of β3 and αIIb was cloned into pcDNA3 and pCEP4 vectors, respectively, as previously described.6 C490A or C545A mutations in αIIb were made by using Site-directed Ligase-Independent Mutagenesis and confirmed by sequencing. Vectors were linearized by using AvrII for αIIb-pCEP4 and PVUI for β3-pcDNA3, then underwent isopropanol clean-up before transfection. BHK cells from American Type Culture Collection maintained in Dulbecco’s modified Eagle medium with 10% FBS, GlutaMAX, and 1% penicillin-streptomycin solution were cotransfected with either wild-type (WT) or C490A or C545A mutant αIIb-pCEP4 vector and WT β3-pcDNA3 vector using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. Antibiotic selection of transfected cells was initiated 48 hours after transfection and maintained in culture. Cells were checked for αIIbβ3 expression by flow cytometry 1-week posttransfection and then sorted based on CD61 expression (clone VI-PL2, Thermo Fisher Scientific). At passaging, cells were washed in PBS and overlayed with 5 mM EDTA in PBS or TrypLE (Gibco) until detached. Cells were then resuspended in fresh media and seeded into a new flask. Cells were used for analysis between passages 3 and 20.
Flow cytometry
BHK cells were washed with PBS and overlayed with 5 mM EDTA in PBS or TrypLE until detached. Cells were counted by using a Countess cell counter and resuspended in ice-cold Hanks’ Balanced Salt Solution (HBSS) containing 5% FBS, 0.1% sodium azide, 1 mM MgCl2, and 1 mM CaCl2. Cells were stained with antibody (diluted in the same buffer) for 30 minutes on ice and washed 3 times with HBSS by centrifugation at 300g for 3 minutes and aspiration of the supernatant. Before acquisition, 0.5 µg/mL 4′,6-diamidino-2-phenylindole (DAPI) was added and cells filtered to remove clumps. Isolated platelets resuspended in HEPES Tyrode’s buffer (20 mM HEPES, 137 mM NaCl, 4 mM KCl, 0.5 mM Na2HPO4, 0.1 mM CaCl2, 5 mM glucose, pH 7.4) were incubated with antibody in a total volume of 100 µL for 20 minutes at 22°C in the dark. Platelets were then fixed by the addition of 200 µL PAMFix (Platelet Solutions) for 5 minutes at 22°C, diluted in 3 mL of HEPES Tyrode’s buffer, and collected by centrifugation at 1500g for 8 minutes with medium acceleration and deceleration. The buffer was removed with a swift upending of the tube, pressing the rim against a tissue, and the platelet pellet resuspended in 500 µL HEPES Tyrode’s buffer. The platelets or BHK cells were acquired on a BD LSR Fortessa analyzer. Antibodies used are as follows: β3 integrin (CD61) was stained with clone VI‐PL2 monoclonal antibody (mAb) conjugated to either allophycocyanin or fluorescein isothiocyanate (FITC) (eBioscience); P-selectin (CD62p) was stained with clone Psel.KO2.3 mAb conjugated to phycoerythrin (eBioscience); and PAC‐1 mAb conjugated to FITC was used to stain for activated αIIbβ3 (BD). In some instances, cells were incubated with 1 µM FITC-labeled fibrinogen (Molecular Innovations) without or with 1 mM Mn2+ in the presence of 20 µM of the αV inhibitor cilengitide (MilliporeSigma) and incubated for 30 minutes at 22°C while agitating.
Cell adhesion
Cells (30 000 per well) were seeded onto a Nunc MaxiSorp flat-bottom 96-well plates (BioLegend) that were coated with 0.1 mg/mL human fibrinogen for 1 hour at 37 °C and blocked with 20 mg/mL bovine serum albumin (BSA) for 1 hour at 37°C. On some occasions, cells were plated in the presence of the αV inhibitor cilengitide at 12.5 µg/mL (MilliporeSigma) and 1 mM manganese sodium salt. Cells were allowed to adhere for 1 hour at 37°C and 5% CO2, after which supernatant was removed and the wells washed 3 times with 200 µL PBS.
Fluorescence microscopy
BHK cells expressing WT β3 integrin and either WT or C490A or C545A mutant αIIb integrin were washed in PBS and overlayed with 5 mM EDTA in PBS for 5 minutes. Cells were de-adhered and resuspended in PBS, counted on a Countess Automatic Cell Counter and then collected by centrifugation at 300g for 10 minutes. Cells were resuspended in HBSS containing 1 mM CaCl2 and 1 mM MgCl2 at 2.5 × 105 cells per mL. Cells were incubated with 0.12 µg/mL FITC- or allophycocyanin-labeled CD61 mAb (clone VI-PL2; Thermo Fisher Scientific) in the presence of 12.5 µg/mL cilengitide for 10 minutes on ice to block αV integrins. Where indicated, cells were collected by centrifugation and resuspended in HBBS containing 0.45 M sucrose and incubated at 22°C for 20 minutes to inhibit receptor internalization before staining. CellCarrier Ultra 96-well plates (Perkin Elmer) were coated with 100 µg/mL fibrinogen for 1 hour at 37°C and blocked with 20 mg/mL BSA for 1 hour at 37°C or coated in 0.1 mg/mL poly-D-lysine (Gibco) and washed in PBS and dried before use. Cells (100 µL per well) were added to wells and allowed to adhere for 1 hour at 37°C and 5% CO2. Unbound cells were removed by washing with PBS and adhered cells fixed with 4% paraformaldehyde for 15 minutes. After washing in PBS, fixed cells were blocked and permeabilized in PBS containing 5% BSA and 0.5% Triton-X100 for 15 minutes. Cells were stained with DAPI and Phalloidin-iFluor 647 (Abcam) and, in some cases, antivinculin eFluor 570 mAb (clone 7F9; Thermo Fischer) or phosphoY397 focal adhesion kinase (FAK) antibody (clone EP216Y; Abcam) in PBS containing 5% BSA and 0.5% Triton-X100 for 20 minutes. Labeled cells were washed in PBS, overlaid in PBS, and imaged by using a PerkinElmer Opera Phenix High-Throughput Imaging System in confocal mode at ×63 magnification. Numerical aperture of the objective used was 1.15 (part no. HH14000342) and camera specifications are 16 bit sCMOS 4.7 Megapixel (2160 × 2160), 6.5 μm pixel size. Data were analyzed by using Harmony version 4.9 software (PerkinElmer).
AP2 assay
BHK cells expressing WT or Cys mutant αIIbβ3 integrin were adhered to a T75 tissue culture flask coated in 0.1 mg/mL fibrinogen for 1 hour at 37°C and blocked in 20 mg/mL BSA for 1 hour at 37°C. After cells had attached, the plate was washed in pre-warmed PBS and the adhered cells overlayed in 1 mL lysis buffer (50 mM Tris pH 7.4 buffer containing 1% triton X-100, 150 mM NaCl, and 0.5 mM EDTA) containing a proteinase inhibitor cocktail (cOmplete Mini EDTA-free Protease Inhibitor cocktail, PhosStop Phosphatase Inhibitor, and 1 mM phenylmethylsulfonyl fluoride). The lysate was diluted twofold in wash buffer (50 mM Tris pH 7.4 buffer containing 0.1% Triton X-100, 150 mM NaCl, and 1 mM EDTA) containing a proteinase inhibitor cocktail (cOmplete Mini EDTA-free Protease Inhibitor cocktail, PhosStop Phosphatase Inhibitor, and 1 mM phenylmethylsulfonyl fluoride). The diluted lysate was incubated with 50 µL of resuspended Protein G Dynabeads (Invitrogen) coupled with 10 µg CD61 mAb according to vendor’s instructions and incubated for 1 hour at 22°C with rotation, after which the beads were washed 3 times in lysis buffer and 3 times in PBS. Captured protein was eluted from the beads by incubation with 30 µL LDS sample buffer and heating at 75°C for 15 minutes. Samples were separated by NuPAGE Novex 4% to 12% Bis-Tris gel with MOPS running buffer and transferred to polyvinylidene fluoride membrane. Membranes were blocked with 5% skim milk in lysis buffer and stained with rabbit polyclonal antibody AP2M1 (ab137727; Abcam) for 60 minutes. After 3 washes in dilution buffer, the blot was stained with peroxidase-conjugated anti-rabbit antibody and the blot imaged. Membranes were reprobed for αIIb (anti-CD41 mAb clone EPR433), and expression of AP2 was normalized to αIIb.
Molecular dynamics simulations
Simulations were prepared from the published integrin αIIbβ3 crystal structure (Protein Data Bank [PDB] identifier 3FCS)7 using the Schrödinger Suite v2020.2 in Schrödinger Maestro v12.4.8 Epik9,10 was used to define protonation states at pH 7 ± 2.0, all crystallographic waters were removed, and all disulfide bonds and bonds to metals were retained. Missing loops and sidechains were repaired with Prime.11 The Thigh domain, Genu, and Calf-1 domain were extracted from the rebuilt structure (residues P452 to R743) and used to model the extension of the integrin headpiece. Termini were capped with N-terminal acetyl and C-terminal N-methylamine functional groups.
All molecular dynamics simulations were performed with the GROMACS 2020.1 engine,12–14 using parameters from the AMBER ff14SB force field15 and a 2-fs time step. The following procedure was applied to 2 systems, 1 in which all 4 disulfide bonds in the domains of interest (C473-C484, C490-C545, C602-C608, and C674-C687) were formed, and 1 in which all but the C490-C545 was formed. These systems were prepared in triplicate by centering the integrin structure at position 7.5 nm, 5 nm, and 12.5 nm in a cubic box with dimensions 15 nm by 20 nm by 12.5 nm and aligning it along the principal protein axis. All simulations were performed under periodic boundary conditions. Systems were solvated in TIP3P water,16 and sufficient ions were added to both neutralize the system charge and to achieve a physiological salt concentration of 150 mM. These configurations were relaxed with steepest-descent energy minimization using at most 50 000 steps.
The minimized structures were thermalized to 300 K over 100 ps under NVT with heavy atom restraints of 1000 kJ/mol nm2 applied to the protein structure. Constant temperature was maintained by using the Bussi velocity rescaling thermostat17 with a coupling constant of 0.1 ps. Heavy-atom–hydrogen-bond vibrations were replaced with holonomic constraints using the LINCS algorithm.18 The smooth Particle Mesh Ewald method19 was used to calculate electrostatic interactions using a cutoff of 1 nm. Simulation seeds were randomized to avoid synchronization artifacts.20 Default values were used for all other parameters. The thermalized systems were then simulated under isothermal-isobaric ensemble (NPT) over 2 ns to ensure equilibration of the periodic box volume. Constant pressure was maintained isotopically at 1 bar by using the Berendsen barostat21 with a coupling constant of 0.2 ps. All other parameters were unchanged from the first equilibration.
The extension of the integrin structure was described with one collective variable, the interior angle between the centers of masses of selected residues within the N-terminal end of the Thigh domain, the Genu, and the C-terminal end of the Calf-1 domain. The residues used to define these centers are listed in supplemental Table 2. Constant-force pulling was applied to the reaction coordinate defined by this collective variable using an umbrella potential with a rate of 0.1 degrees per ps and a force constant of 1000 kJ/mol radian2. The force induced under these conditions was deemed appropriate on the basis of the resulting trajectories, as protein deformation was not observed, and the forces observed in previous simulations by Zhu et al.7 Pulling was conducted over 200 ps, with heavy atom restraints of 1000 kJ mol-1 nm-2applied to the center of mass of selected residues within the N-terminus of the Thigh domain; this center of mass was used as an immobile reference for the pulling simulations. Trajectories were saved every 2 ps. All other parameters were unchanged from the second equilibration.
Calculation of the free energy landscape for the extension of the integrin headpiece was conducted by using umbrella sampling. Thirty sampling windows were selected from each of the previously described pulling simulations such that the window spacing was ∼2.5 degrees. After velocities were discarded, each window was equilibrated under NPT for 200 ps. Constant pressure was maintained isotopically at 1 bar by using the Parinello-Rahman barostat22 with a coupling constant of 5 ps. The initial angle of each sampling window was maintained with an umbrella potential throughout equilibration and production. All other parameters were unchanged. Each window was then subjected to 50 ns of simulation under a harmonic potential, from which data were collected every 10 000 steps, for a total of 1.5 µsec per replicate. Construction of the free energy landscape was performed with the weighted histogram analysis method module within GROMACS 2020.2.23 Minima in the resultant potential of mean force curves were matched at an angle of ∼120 degrees. Errors were estimated by Bayesian bootstrapping (10 000 iterations) of complete autocorrelated trajectories from the combined umbrella histograms of each system, using the weighted histogram analysis method module. Autocorrelation times for each window were found to be significantly smaller than the sampling time, being one order of magnitude smaller in the worst case, and multiple independent histograms overlap in each sampling window, such that the error estimates are deemed reliable.
Force distribution analysis of molecular dynamics simulations was performed with the GROMACS–force distribution analysis module v2.1024 and used to provide insight into mechanical signal propagation within the integrin structure with an intact or missing C490-C545 bond. The pairwise forces between protein residues were calculated at every frame, whereas those from water molecules and metal ions were neglected. The time-averaged force was computed (<Fij(reduced)> – <Fij(oxidized)>) and visualized by using PyMOL and are shown if greater than the specified threshold values.
Principal component analysis was performed by using MDTraj425 and scikit-learn.26 The concatenated trajectories of the backbone atoms for the integrin structure with an intact or missing C490-C545 bond were used as a common basis after least squares fitting; the concatenated trajectory of each redox state was then individually projected along the first 2 eigenvectors of the covariance matrix, which account for ∼22% of the explained variance in each case.
Results
The αIIb C490-C545 disulfide bond is missing in approximately one-third of integrin molecules on the resting and activated human platelet surface
The redox state of cysteine thiols in the αIIb subunit of platelet αIIbβ3 integrin was determined by using differential cysteine alkylation with a pair of isotopic alkylators and mass spectrometry.27 The 18 cysteines in the αIIb subunit participate in 9 disulfide bonds that have been structurally defined in an X-ray structure of the extracellular region of resting αIIbβ3 integrin (PDB identifier 3fcs7) (Figure 1A).
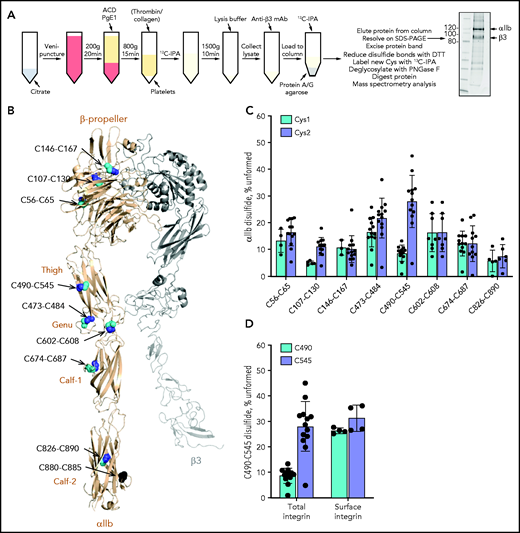
The αIIb C490-C545 disulfide bond is missing in approximately one-third of integrin molecules on the resting and activated human platelet surface. (A) Blood from healthy volunteers was drawn by venipuncture into 0.4% citrate as an anticoagulant. Platelet-rich plasma was prepared by centrifugation and transferred into a new tube with 20% (v/v) acid citrate dextrose (ACD) solution. After a 30-minute rest at 37°C, 1 µM prostaglandin E1 (PgE1) was added and platelets pelleted by centrifugation. The platelet pellet was resuspended in warm HEPES Tyrode’s buffer and unpaired cysteine thiols alkylated by the addition of 4 mM 12C-IPA, in some cases after the addition of 1 U/mL thrombin and 5 µg/mL collagen to activate the platelets. Labeled platelets were pelleted and resuspended in lysis buffer. The lysate was diluted in HEPES-buffered saline and incubated with anti-β3 mAb; the antibody-labeled integrin was then collected on protein A/G agarose, washed, labeled for a second time with 4 mM 12C-IPA, eluted in LDS buffer, and resolved on SDS-PAGE. The αIIb band was excised and washed before disulfide-bonded cysteines were reduced with dithiothreitol (DTT), and the resulting new cysteine thiols labeled with 5 mM 13C-IPA. The protein was deglycosylated by using PNGase F and digested with chymotrypsin and trypsin. Twenty-three peptides reporting on 8 of the 9 αIIb disulfide bonds were analyzed by using mass spectrometry (Table 1; supplemental Figure 1). The redox state of disulfide bonds was quantified as the percentage of 12C-IPA labeled Cys in which the total was the sum of 12C-IPA– and 13C-IPA–labeled Cys. Example of a Sypro Ruby–stained gel of 12C-IPA–labeled αIIbβ3 resolved on SDS-PAGE is shown at right. Molecular mass standards are shown in the left lane. (B) Positions of the αIIb integrin disulfide bonds in a modeled open structure4 of the complete αIIbβ3 integrin ectodomain (αIIb subunit in wheat ribbon and β3 subunit in gray ribbon). The cysteine residues comprising the nine αIIb disulfide bonds are shown as cyan, blue, and black spheres and the residue numbers indicated. The redox state of the cysteines in cyan and blue was measured by using differential alkylation and by mass spectrometry. The cysteines in black (C880 and C885) were not determined. Domain names are indicated. (C) Redox states of eight αIIb disulfide bonds in 13 healthy human donor platelets (6 male subjects, 7 female subjects; 21-48 years old). The bars and errors are mean ± standard deviation. (D) Percentage of the αIIb C490-C545 disulfide bond that is missing in total (6 male subjects, 7 female subjects; 21-48 years old) and platelet surface (2 female subjects; 22 and 23 years old, biological replicates) integrin in healthy human donor platelets. The bars and errors are mean ± standard deviation.
The αIIb C490-C545 disulfide bond is missing in approximately one-third of integrin molecules on the resting and activated human platelet surface. (A) Blood from healthy volunteers was drawn by venipuncture into 0.4% citrate as an anticoagulant. Platelet-rich plasma was prepared by centrifugation and transferred into a new tube with 20% (v/v) acid citrate dextrose (ACD) solution. After a 30-minute rest at 37°C, 1 µM prostaglandin E1 (PgE1) was added and platelets pelleted by centrifugation. The platelet pellet was resuspended in warm HEPES Tyrode’s buffer and unpaired cysteine thiols alkylated by the addition of 4 mM 12C-IPA, in some cases after the addition of 1 U/mL thrombin and 5 µg/mL collagen to activate the platelets. Labeled platelets were pelleted and resuspended in lysis buffer. The lysate was diluted in HEPES-buffered saline and incubated with anti-β3 mAb; the antibody-labeled integrin was then collected on protein A/G agarose, washed, labeled for a second time with 4 mM 12C-IPA, eluted in LDS buffer, and resolved on SDS-PAGE. The αIIb band was excised and washed before disulfide-bonded cysteines were reduced with dithiothreitol (DTT), and the resulting new cysteine thiols labeled with 5 mM 13C-IPA. The protein was deglycosylated by using PNGase F and digested with chymotrypsin and trypsin. Twenty-three peptides reporting on 8 of the 9 αIIb disulfide bonds were analyzed by using mass spectrometry (Table 1; supplemental Figure 1). The redox state of disulfide bonds was quantified as the percentage of 12C-IPA labeled Cys in which the total was the sum of 12C-IPA– and 13C-IPA–labeled Cys. Example of a Sypro Ruby–stained gel of 12C-IPA–labeled αIIbβ3 resolved on SDS-PAGE is shown at right. Molecular mass standards are shown in the left lane. (B) Positions of the αIIb integrin disulfide bonds in a modeled open structure4 of the complete αIIbβ3 integrin ectodomain (αIIb subunit in wheat ribbon and β3 subunit in gray ribbon). The cysteine residues comprising the nine αIIb disulfide bonds are shown as cyan, blue, and black spheres and the residue numbers indicated. The redox state of the cysteines in cyan and blue was measured by using differential alkylation and by mass spectrometry. The cysteines in black (C880 and C885) were not determined. Domain names are indicated. (C) Redox states of eight αIIb disulfide bonds in 13 healthy human donor platelets (6 male subjects, 7 female subjects; 21-48 years old). The bars and errors are mean ± standard deviation. (D) Percentage of the αIIb C490-C545 disulfide bond that is missing in total (6 male subjects, 7 female subjects; 21-48 years old) and platelet surface (2 female subjects; 22 and 23 years old, biological replicates) integrin in healthy human donor platelets. The bars and errors are mean ± standard deviation.
Briefly, blood from healthy donors was drawn into citrate as an anticoagulant, platelets prepared by centrifugation, and unpaired cysteine thiols in platelet proteins alkylated with a 12C isotope of IPA (12C-IPA). The platelets were lysed, αIIbβ3 integrin collected on antibody-coated magnetic beads, and resolved on SDS-PAGE (Figure 1A). The αIIb subunit was excised from the gel, reduced with dithiothreitol, and the disulfide-bonded cysteine thiols alkylated with a 13C isotope of IPA (13C-IPA).28 The protein was digested with trypsin and chymotrypsin, peptides quantified by using high-performance liquid chromatography, and identity established by mass spectrometry (Table 1; supplemental Figure 1). The levels of the different redox forms of the αIIb cysteines were determined from the relative abundance of peptides labeled with 12C-IPA and/or 13C-IPA. The results are expressed as a percentage of the αIIb disulfide that is missing in the population. We have used the term “missing” disulfide bond, as opposed to “reduced” disulfide bond, as reduced implies that the bond was formed at some point and later reduced, for which there is no evidence.
αIIb subunit peptides analyzed to determine the disulfide bond redox state
| Disulfide bond | Cysteine | Peptide sequence* | Peptide score | Peptide error (ppm) |
|---|---|---|---|---|
| C56-C65 | C56 | LCPWR | 196† | –0.5 |
| C65 | RAEGGQCPSLL | 39 | –5.8 | |
| RAEGGQCPSLLF | 43 | 1.7 | ||
| C107-C130 | C107 | SDVIVACAPW | 286† | –2.4 |
| C130 | TEEAEKTPVGSCF | 68 | –3.5 | |
| EKTEEAEKTPVGSCF | 74 | 1.9 | ||
| TPVGSCF | 429† | –2.6 | ||
| C146-C167 | C146 | AEYSPCR | 38 | –3.4 |
| C167 | CEAGF | 433† | –2.9 | |
| C473-C484 | C473 | SCVLPQTK | 257† | –1.5 |
| C484 | TPVSCF | 432† | 4.4 | |
| TKTPVSCF | 451† | –1.2 | ||
| C490-C545 | C490 | NIQMCVGATGH | 379† | –2.6 |
| NIQM(oxidation)CVGATGH | 335† | –2.6 | ||
| C545 | HSPICHTTM | 40 | –2.1 | |
| HSPICHTTM(oxidation) | 284† | –0.3 | ||
| HSPICH | 410† | –2.0 | ||
| C602-C608 | C602, C608 | DCGEDDVCVPQLQL | 247† | –2.2 |
| C674-C687 | C674 | ICNQK | 375† | –3.3 |
| LICNQK | 284† | –2.5 | ||
| C687 | CELGNPM(oxidation)K | 341† | –2.5 | |
| CELGNPM(oxidation)KK | 41 | –2.4 | ||
| C826-C890 | C826 | QCFPQPPVNPL | 447† | 0.1 |
| C890 | CDLQEM(oxidation)AR | 286† | 0.8 |
| Disulfide bond | Cysteine | Peptide sequence* | Peptide score | Peptide error (ppm) |
|---|---|---|---|---|
| C56-C65 | C56 | LCPWR | 196† | –0.5 |
| C65 | RAEGGQCPSLL | 39 | –5.8 | |
| RAEGGQCPSLLF | 43 | 1.7 | ||
| C107-C130 | C107 | SDVIVACAPW | 286† | –2.4 |
| C130 | TEEAEKTPVGSCF | 68 | –3.5 | |
| EKTEEAEKTPVGSCF | 74 | 1.9 | ||
| TPVGSCF | 429† | –2.6 | ||
| C146-C167 | C146 | AEYSPCR | 38 | –3.4 |
| C167 | CEAGF | 433† | –2.9 | |
| C473-C484 | C473 | SCVLPQTK | 257† | –1.5 |
| C484 | TPVSCF | 432† | 4.4 | |
| TKTPVSCF | 451† | –1.2 | ||
| C490-C545 | C490 | NIQMCVGATGH | 379† | –2.6 |
| NIQM(oxidation)CVGATGH | 335† | –2.6 | ||
| C545 | HSPICHTTM | 40 | –2.1 | |
| HSPICHTTM(oxidation) | 284† | –0.3 | ||
| HSPICH | 410† | –2.0 | ||
| C602-C608 | C602, C608 | DCGEDDVCVPQLQL | 247† | –2.2 |
| C674-C687 | C674 | ICNQK | 375† | –3.3 |
| LICNQK | 284† | –2.5 | ||
| C687 | CELGNPM(oxidation)K | 341† | –2.5 | |
| CELGNPM(oxidation)KK | 41 | –2.4 | ||
| C826-C890 | C826 | QCFPQPPVNPL | 447† | 0.1 |
| C890 | CDLQEM(oxidation)AR | 286† | 0.8 |
Peptides were detected by using both Byonic and Mascot analysis software, confirmed by using tandem mass spectrometry, and have errors <6 ppm. Only peptides with peak areas >3 million and contributing >10% of the total peptide peak areas for a given Cys were included in the analysis.
Cys (underlined) was labeled with 12C-iodoacetanilide or 13C-iodoacetanilide.
Score and error were determined by using Byonic software using αIIb integrin as reference.
We were able to quantify the redox state of 8 of the 9 disulfide bonds in αIIb (Figure 1B). The exception was the C880-C885 disulfide in the Calf-2 domain. The redox state of both cysteines for all 8 disulfide bonds (C56-C65, C107-C130, and C146-C167 in the β-propeller domain; C490-C545 and C473-C484 in the Thigh domain; C602-C608 in the Genu domain; C674-C687 in the Calf-1 domain; and C826-C890 in the Calf-2 domain) was measured. Seven of the 8 disulfide bonds in platelet αIIb were more than ∼80% formed in the platelet integrin populations of 13 healthy human donors (6 male subjects, 7 female subjects; 21-48 years old) (Figure 1C). The exception was the C490-C545 bond in the Thigh domain that is missing in 28% (range, 5%-45%) of the integrin subunit based on the redox state of C545. In other words, the C490-C545 bond is missing in ∼1 in 3 molecules on the platelet surface. There was donor-to-donor variation in the redox state of the C490-C545 bond with a coefficient of variation of 35%. The donor variation was not due to assay variation, as repeated measurements of the C490-C545 disulfide state in 3 different donors showed very small interassay variation (supplemental Figure 2). There was no significant age or sex difference in the data cohort (supplemental Figure 3).
It was unexpected that C545 is ∼30% in free thiol form in human platelet αIIb, whereas the cysteine that it pairs with, C490, is ∼10% in free thiol form (Figure 1C). The reason for this difference has been investigated and is discussed in the supplemental Materials. The incidence of the integrin state in the total platelet pool reflects the incidence on the platelet surface (Figure 1D). Notably, 12C-IPA alkylation of C490 is comparable to the level of alkylation of C545 when only platelet surface integrin was analyzed, indicating that the reduced alkylation of C490 is a particular feature of the intraplatelet pool of the integrin.
An important question is whether αIIb arrives on the platelet surface with a missing C490-C545 disulfide bond, or newly formed platelets contain integrin with a formed C490-C545 disulfide bond that is cleaved in the circulation.
Redox state of the C490-C545 disulfide bond in αIIbβ3 integrin is predetermined and not a result of postsecretion redox changes in the blood
Strong activation of washed platelets with thrombin and collagen had no effect on the redox state of the C490-C545 disulfide bond (Figure 2A), indicating that platelet activation status is not involved in the redox state of the αIIb bond. Platelet activation in this experiment was confirmed by surface expression of P-selectin and activated αIIbβ3 integrin (supplemental Figure 6).

αIIbβ3 integrin with a missing αIIb C490-C545 disulfide bond is predetermined and selectively partitions into focal adhesions on the activated platelet surface. The result for C545 for the total integrin pool is shown as it represents the fraction of the C490-C545 disulfide bond that is unformed (as discussed in Figure 1). (A) Redox state of the αIIb C490-C545 disulfide bond in healthy human donor resting platelets (6 male subjects, 7 female subjects; 21-48 years old), thrombin/collagen–activated platelets (5 male subjects, 4 female subjects; 23-48 years old), αIIbβ3 naturally produced by human megakaryoblastic MEG-01 cells (4 different preparations), and recombinantly expressed in mammalian BHK cells (3 independent experiments). The bars and errors are mean ± standard deviation. (B) Redox state of the αIIb C490-C545 disulfide bond in the total integrin pool or bound to talin in focal adhesions in resting or thrombin-activated donor platelets (2 male subjects, 1 female subject; 29-42 years old). The bars and errors are mean ± standard deviation. *P < .05.
αIIbβ3 integrin with a missing αIIb C490-C545 disulfide bond is predetermined and selectively partitions into focal adhesions on the activated platelet surface. The result for C545 for the total integrin pool is shown as it represents the fraction of the C490-C545 disulfide bond that is unformed (as discussed in Figure 1). (A) Redox state of the αIIb C490-C545 disulfide bond in healthy human donor resting platelets (6 male subjects, 7 female subjects; 21-48 years old), thrombin/collagen–activated platelets (5 male subjects, 4 female subjects; 23-48 years old), αIIbβ3 naturally produced by human megakaryoblastic MEG-01 cells (4 different preparations), and recombinantly expressed in mammalian BHK cells (3 independent experiments). The bars and errors are mean ± standard deviation. (B) Redox state of the αIIb C490-C545 disulfide bond in the total integrin pool or bound to talin in focal adhesions in resting or thrombin-activated donor platelets (2 male subjects, 1 female subject; 29-42 years old). The bars and errors are mean ± standard deviation. *P < .05.
Platelets are anucleate cells derived from the cytoplasm of megakaryocytes residing in the bone marrow. Integrins, along with granules and other organelle content, are packaged into proplatelets that are released into the bloodstream and form platelets.29 The redox state of the C490-C545 disulfide bond was assessed in a human megakaryoblastic cell line, MEG-01. The αIIb integrin produced by MEG-01 cells has the same redox state as the platelet integrin; the C490-C545 disulfide bond is missing in 32 ± 7% of the subunit (Figure 2A). This result implies that ∼1 in 3 integrin molecules produced by megakaryocytes contain a missing C490-C545 disulfide bond, which is then sorted into blood platelets at the same ratio.
We also measured the redox state of the C490-C545 bond in recombinant WT αIIbβ3 integrin expressed in BHK cells to test whether the same αIIb subset is produced by a mammalian cell unrelated to platelet production. The recombinant αIIb integrin produced by BHK cells has the same redox state as the platelet and MEG-01 integrin; the C490-C545 disulfide bond is missing in 43 ± 14% of the subunit (Figure 2A). In addition, the C490-C545 disulfide in purified αIIbβ3 integrin is not cleaved by any of the known vascular thiol isomerases: PDI, ERp5, ERp57, ERp72, and TMX130,31 (supplemental Figure 7). These results indicate that the integrin state is predetermined and not a result of postsecretion modifications of an oxidized integrin at the platelet surface.
We next determined if the alternate disulfide-bonded form of αIIbβ3 integrin selectively partitions into different areas of the platelet surface. We chose to examine distribution of the alternate form in focal adhesions in activated platelets. Focal adhesions are multiprotein complexes at the plasma membrane that coordinate mechanical and regulatory signals between the cell and its environment.
αIIbβ3 integrin with a missing C490-C545 disulfide bond selectively partitions into focal adhesions on the activated platelet surface
Talin binds to the cytoplasmic face of integrins in focal adhesions. The redox state of the αIIb C490-C545 disulfide bond in the total integrin pool compared with that bound to talin in focal adhesions in resting and thrombin-activated donor platelets was determined. The immunoprecipitation was performed by using talin mAb, and the αIIb that coimmunoprecipitated with talin was analyzed. In resting platelets that contain very few focal adhesions, there was no difference in the distribution of the alternate covalent form between the total integrin pool and bound to talin. In contrast, in thrombin-activated platelets, the distribution of the alternate disulfide-bonded form of αIIbβ3 shifted from the total pool to focal adhesions (P < .05) (Figure 2B). This result indicates that αIIbβ3 integrin with a missing C490-C545 disulfide bond selectively partitions into focal adhesions on the activated platelet surface.
The functional consequences of a missing C490-C545 disulfide bond for integrin function were evaluated in mammalian cells expressing a mutant integrin with an ablated bond.
Ablation of the αIIb C490-C545 disulfide bond restricts the distribution of αIIbβ3 to focal adhesions
Cysteine to alanine mutations of C490 and C545 were made and expressed together with WT β3 in mammalian BHK cells. The cells were sorted on β3 expression to achieve lines with comparable expression of WT and C490A and C545A mutant αIIbβ3 integrins (Figure 3A). The double C490, C545A mutant was not expressed at the cell surface in BHK cells.

Ablation of the αIIb C490-C545 disulfide bond restricts the distribution of αIIbβ3 to focal adhesions. (A) BHK cells were transfected with WT β3 and either WT or cysteine mutant αIIb and cells sorted for high β3 expression. Expression of β3 in the clones used for the experiments is shown as a bar graph (i) or representative histogram (ii). The bars and errors are mean ± standard error of the mean of 6 biological replicates. (B) Ablation of the αIIb C490-C545 disulfide bond reduces adhesion of cells to immobilized fibrinogen but not poly-D-lysine. The bars and errors are mean ± standard deviation (SD) of 4 independent experiments with 1 or 2 biological replicates. (C) WT or cysteine mutant αIIb-expressing BHK cells were seeded onto a fibrinogen-coated surface in the presence of β3 mAb VI-PL2 and allowed to adhere for 1 hour. After fixation, cells were stained for vinculin (7F9) and nuclei (DAPI), before being imaged using PerkinElmer’s Opera Phenix High-Content Screening System at ×63 magnification in confocal mode. Scale bar, 50 µm. (D) Integrin clusters were quantified in cells seeded onto poly-D-lysine or fibrinogen and expressed per cell (i) or per square micrometers (ii). Discreet integrin clusters in cells adhered to fibrinogen were more abundant when the αIIb C490-C545 disulfide bond is missing. The bars and errors are mean ± SD of 4 to 8 biological replicates. (E) WT or cysteine mutant αIIb-expressing BHK cells were seeded onto a fibrinogen-coated surface in the presence of β3 mAb VI-PL2 and allowed to adhere for 1 hour. After fixation, cells were stained for vinculin (7F9), phosphorylated FAK (pFAK, phosphoY397 FAK mAb), and nuclei (DAPI ), before being imaged by using PerkinElmer’s Opera Phenix high-content screening system at ×63 magnification in confocal mode. Scale bar, 50 µm. (F) F-actin and pFAK intensity in cells seeded on the fibrinogen-coated surface are expressed per µm2. The bars and errors are mean ± SD of 3 to 8 biological replicates. *P < .05, **P < .01, ****P < .0001 (1-way analysis of variance).
Ablation of the αIIb C490-C545 disulfide bond restricts the distribution of αIIbβ3 to focal adhesions. (A) BHK cells were transfected with WT β3 and either WT or cysteine mutant αIIb and cells sorted for high β3 expression. Expression of β3 in the clones used for the experiments is shown as a bar graph (i) or representative histogram (ii). The bars and errors are mean ± standard error of the mean of 6 biological replicates. (B) Ablation of the αIIb C490-C545 disulfide bond reduces adhesion of cells to immobilized fibrinogen but not poly-D-lysine. The bars and errors are mean ± standard deviation (SD) of 4 independent experiments with 1 or 2 biological replicates. (C) WT or cysteine mutant αIIb-expressing BHK cells were seeded onto a fibrinogen-coated surface in the presence of β3 mAb VI-PL2 and allowed to adhere for 1 hour. After fixation, cells were stained for vinculin (7F9) and nuclei (DAPI), before being imaged using PerkinElmer’s Opera Phenix High-Content Screening System at ×63 magnification in confocal mode. Scale bar, 50 µm. (D) Integrin clusters were quantified in cells seeded onto poly-D-lysine or fibrinogen and expressed per cell (i) or per square micrometers (ii). Discreet integrin clusters in cells adhered to fibrinogen were more abundant when the αIIb C490-C545 disulfide bond is missing. The bars and errors are mean ± SD of 4 to 8 biological replicates. (E) WT or cysteine mutant αIIb-expressing BHK cells were seeded onto a fibrinogen-coated surface in the presence of β3 mAb VI-PL2 and allowed to adhere for 1 hour. After fixation, cells were stained for vinculin (7F9), phosphorylated FAK (pFAK, phosphoY397 FAK mAb), and nuclei (DAPI ), before being imaged by using PerkinElmer’s Opera Phenix high-content screening system at ×63 magnification in confocal mode. Scale bar, 50 µm. (F) F-actin and pFAK intensity in cells seeded on the fibrinogen-coated surface are expressed per µm2. The bars and errors are mean ± SD of 3 to 8 biological replicates. *P < .05, **P < .01, ****P < .0001 (1-way analysis of variance).
Platelet plug formation at sites of blood vessel injury is mediated by cross-linking of αIIbβ3 integrin by the bivalent ligand fibrinogen. Mutation of the αIIb C490-C545 disulfide bond reduced BHK cell adhesion to immobilized fibrinogen but not poly-D-lysine (P < .01) (Figure 3B), indicating that replacing C490 or C545 with alanine reduces but does not ablate binding to fibrinogen. The BHK cells were adhered to a fibrinogen-coated surface and imaged by confocal immunofluorescence (Figure 3C). The plasma membrane distribution of integrin is clearly different for WT and disulfide mutant. WT integrin is distributed both within and without focal adhesions (indicated by focal adhesion protein vinculin) in the plasma membrane, whereas ablation of the αIIb C490-C545 disulfide bond results in restriction of the integrin mostly to focal adhesions. The number of integrin clusters on the surface of αIIb cysteine mutant cells is significantly higher than in WT integrin cells (P < .0001) (Figure 3D), which was not the case when cells were adhered to control poly-D-lysine.
Platelet αIIbβ3 activation and clustering are necessary for outside-in signaling32 that involves downstream phosphorylation of FAK.33 Filamentous actin (F-actin) is formed after integrin activation and is required for platelet shape change.34 FAK phosphorylation and F-actin formation in response to fibrinogen binding in BHK cells expressing WT or C490A and C545A mutant αIIbβ3 integrins were measured by confocal immunofluorescence of cells seeded on a fibrinogen-coated surface. Both FAK phosphorylation and F-actin formation were comparable in cells expressing WT or disulfide mutant αIIbβ3 integrins (Figure 3E-F) when difference in cell spread size is considered (Figure 4A), indicating that the alternate covalent form of αIIbβ3 has functional outside-in signaling.

Ablation of the αIIb C490-C545 disulfide bond extends residency time of αIIbβ3 integrin in focal adhesions due to a reduced rate of clathrin-mediated integrin internalization and recycling. (A) WT or cysteine mutant αIIβ3-expressing cells were seeded onto a fibrinogen-coated surface for 1 hour, fixed, stained for F-actin intensity, and the cell area measured. The surface area of spread cells is smaller when the αIIb C490-C545 disulfide bond is missing. The bars and errors are mean ± standard deviation (SD) of 8 biological replicates, and each data point represents the mean surface area of at least 500 cells. (B) WT or cysteine mutant αIIβ3-expressing cells were treated with 0.45 M sucrose to inhibit clathrin-mediated endocytosis (CME) before seeding on a fibrinogen-coated surface in the presence of β3 mAb VI-PL2 for 1 hour. After fixation, nuclei were stained with DAPI and spread area measured. Panel i is a representative image of WT cells before and after CME inhibition; panel ii is spread cell area for WT or cysteine mutant αIIβ3-expressing cells. The bars and errors are mean ± SD of 4 biological replicates, and each data point represents the mean surface area of at least 500 cells. (C) Cells stained with β3 mAb VI-PL2 were seeded onto a fibrinogen-coated surface and imaged every 30 seconds for 50 minutes. Integrin clusters were identified and observed. Panel i is the mean β3 cluster lifetime as a function of time for 3 independent experiments for at least 100 cells each; panel ii is the mean and SD of all measurements. (D) WT or cysteine mutant αIIβ3-expressing cells were seeded onto a fibrinogen-coated surface for 1 hour, αIIbβ3 immunoprecipitated from cell lysate, and blotted for αIIb and AP2. Panel i is a representative blot; panel ii includes AP2/αIIb band intensity ratios for 4 biological replicates. The bars and errors are mean ± SD. *P < .05, **P < .01, ****P < .0001 (1-way analysis of variance). ns, not significant.
Ablation of the αIIb C490-C545 disulfide bond extends residency time of αIIbβ3 integrin in focal adhesions due to a reduced rate of clathrin-mediated integrin internalization and recycling. (A) WT or cysteine mutant αIIβ3-expressing cells were seeded onto a fibrinogen-coated surface for 1 hour, fixed, stained for F-actin intensity, and the cell area measured. The surface area of spread cells is smaller when the αIIb C490-C545 disulfide bond is missing. The bars and errors are mean ± standard deviation (SD) of 8 biological replicates, and each data point represents the mean surface area of at least 500 cells. (B) WT or cysteine mutant αIIβ3-expressing cells were treated with 0.45 M sucrose to inhibit clathrin-mediated endocytosis (CME) before seeding on a fibrinogen-coated surface in the presence of β3 mAb VI-PL2 for 1 hour. After fixation, nuclei were stained with DAPI and spread area measured. Panel i is a representative image of WT cells before and after CME inhibition; panel ii is spread cell area for WT or cysteine mutant αIIβ3-expressing cells. The bars and errors are mean ± SD of 4 biological replicates, and each data point represents the mean surface area of at least 500 cells. (C) Cells stained with β3 mAb VI-PL2 were seeded onto a fibrinogen-coated surface and imaged every 30 seconds for 50 minutes. Integrin clusters were identified and observed. Panel i is the mean β3 cluster lifetime as a function of time for 3 independent experiments for at least 100 cells each; panel ii is the mean and SD of all measurements. (D) WT or cysteine mutant αIIβ3-expressing cells were seeded onto a fibrinogen-coated surface for 1 hour, αIIbβ3 immunoprecipitated from cell lysate, and blotted for αIIb and AP2. Panel i is a representative blot; panel ii includes AP2/αIIb band intensity ratios for 4 biological replicates. The bars and errors are mean ± SD. *P < .05, **P < .01, ****P < .0001 (1-way analysis of variance). ns, not significant.
These results indicate that ablation of the αIIb C490-C545 disulfide bond largely restricts the distribution of αIIbβ3 to focal adhesions, in accordance with the distribution of this alternate covalent form of αIIbβ3 in activated human platelets (Figure 2B). Clustering of activated integrins in focal adhesions is a critical step for clathrin-mediated endocytosis and recycling of integrins back to the plasma membrane.33 FAK signaling is also associated with focal adhesion turnover.35 We therefore tested effect of ablation of the αIIb C490-C545 disulfide on integrin internalization or recycling.
Ablation of the αIIb C490-C545 disulfide bond extends residency time of αIIbβ3 integrin in focal adhesions due to reduced rate of clathrin-mediated integrin internalization and recycling
Cells expressing the αIIb cysteine mutants have a significantly smaller surface area than cells expressing WT integrin (P < .0001) (Figure 4A). Clathrin-mediated endocytosis in the BHK cells was inhibited by incubation with a high-sucrose concentration before adherence to fibrinogen. Blocking clathrin-mediated endocytosis in WT or C490A and C545A mutant αIIbβ3 integrin cells mostly negated the change in spread cell size between WT and disulfide mutants (Figure 4B). This result supports a role for the αIIb C490-C545 disulfide bond in integrin internalization/recycling. Rate of turnover of integrin-containing focal adhesions on the surface of the BHK cells was quantified to test this conclusion.
BHK cells were labeled with β3 antibody, adhered to a fibrinogen-coated surface, and imaged every 30 seconds. The integrin clusters that appear on the surface of the cells were tracked and their lifetime plotted over the course of 60 minutes. As anticipated, the lifetime of integrin-rich focal adhesions was significantly longer on cells expressing C490A and C545A mutant αIIbβ3 integrin compared with WT (P < .0001) (Figure 4C).
Internalization of integrins is selectively regulated by the clathrin adaptor protein AP236 that binds to the cytoplasmic domain of αIIbβ3.37 Association of AP2 with αIIbβ3 in BHK cells expressing WT or C490A and C545A mutant αIIbβ3 integrins was measured by coimmunoprecipitation and western blot. There was significantly enhanced binding of AP2 to αIIbβ3 integrin when the C490-C545 disulfide bond is missing (P < .05) (Figure 4D).
Most integrins, including αIIbβ3, are in a quiescent state on the cell surface that does not bind ligand or signal. The receptors are activated by intracellular stimuli such as talin engagement of the cytoplasmic region via a process termed inside-out signaling and by ligand binding to the extracellular region that transmits information to the cytoplasm via outside-in signaling.38,39 Global and local conformational rearrangements of the integrin ectodomains control affinity for ligands. The quiescent integrin exists predominantly in a bent conformation with a closed headpiece. The active fully extended conformation with open headpiece is achieved via at least 6 intermediate conformations.40 Three main conformational states have been defined: bent conformation with closed headpiece (low affinity for ligand), extended conformation with closed headpiece (intermediate affinity), and extended conformation with open headpiece (high affinity). Ablation of the αIIb C490-C545 disulfide bond in BHK cells reduced affinity for soluble fibrinogen and activation of the mutant integrins by manganese ions (P < .01) (Figure 5A), suggesting altered conformational dynamics of the mutant integrin. The reduced rate of clathrin-mediated integrin internalization and recycling and enhanced AP2 binding of the alternate αIIbβ3 integrin form also implies change in integrin conformation. This was examined by molecular dynamics simulations of bending at the αIIb subunit knee joint.

Molecular dynamics simulations show that αIIb integrin with a missing C490-C545 disulfide bond requires higher forces to transition from bent to open conformational states. (A) Ablation of the αIIb C490-C545 disulfide bond in BHK cells reduces integrin activation by manganese. The bars and errors are mean ± standard deviation of 3 independent experiments with 2 biological replicates each. (B) Molecular dynamics simulations were performed on the Thigh, Genu, and Calf-1 domain of αIIb integrin using the crystal structure of “bent” αIIbβ3 integrin was used to model the effect of the intact or missing disulfide bond (PDB identifier 3fcs).7 Conformational distribution of the αIIbβ3 integrin with an intact (top) and missing (bottom) C490-C545 disulfide bond, projected along the first 2 principal components (PC1 and PC2, respectively), supporting the notion that the redox state of the bond has significant structural implications. Conformational density (normalized, arbitrary units) is indicated from low (dark blue) to high (cyan). (C) Difference network of averaged pairwise-forces48 (stick representation), measured between the integrin in both redox states. Force magnitude is indicated by stick color, from blue to green; only forces >25 kJ/mol nm are presented. Significant differences in the force networks are observed in the Genu domain, between the Thigh and Calf-1 domains, despite being distant from the missing disulfide, indicating the propagation of subtle mechanical forces throughout the protein structure. The C490-C545 disulfide is shown in stick representation, and a calcium ion is represented as an orange sphere. (D) Potential of mean force calculations suggest that the closed form of the integrin is disfavored when the C490-C545 bond is missing, reducing cycling efficiency. Representative structures from umbrella sampling are overlaid at 60, 100, and 140 degrees . **P < .01. MFI, mean fluorescence intensity.
Molecular dynamics simulations show that αIIb integrin with a missing C490-C545 disulfide bond requires higher forces to transition from bent to open conformational states. (A) Ablation of the αIIb C490-C545 disulfide bond in BHK cells reduces integrin activation by manganese. The bars and errors are mean ± standard deviation of 3 independent experiments with 2 biological replicates each. (B) Molecular dynamics simulations were performed on the Thigh, Genu, and Calf-1 domain of αIIb integrin using the crystal structure of “bent” αIIbβ3 integrin was used to model the effect of the intact or missing disulfide bond (PDB identifier 3fcs).7 Conformational distribution of the αIIbβ3 integrin with an intact (top) and missing (bottom) C490-C545 disulfide bond, projected along the first 2 principal components (PC1 and PC2, respectively), supporting the notion that the redox state of the bond has significant structural implications. Conformational density (normalized, arbitrary units) is indicated from low (dark blue) to high (cyan). (C) Difference network of averaged pairwise-forces48 (stick representation), measured between the integrin in both redox states. Force magnitude is indicated by stick color, from blue to green; only forces >25 kJ/mol nm are presented. Significant differences in the force networks are observed in the Genu domain, between the Thigh and Calf-1 domains, despite being distant from the missing disulfide, indicating the propagation of subtle mechanical forces throughout the protein structure. The C490-C545 disulfide is shown in stick representation, and a calcium ion is represented as an orange sphere. (D) Potential of mean force calculations suggest that the closed form of the integrin is disfavored when the C490-C545 bond is missing, reducing cycling efficiency. Representative structures from umbrella sampling are overlaid at 60, 100, and 140 degrees . **P < .01. MFI, mean fluorescence intensity.
Molecular dynamics simulations reveal that ablation of the αIIb C490-C545 disulfide bond changes bending at the knee joint
The crystal structure of resting (bent) αIIbβ3 integrin (PDB identifier 3fcs)7 was used to model the effect of the intact or missing αIIb C490-C545 disulfide bond on extension of the integrin headpiece. Molecular dynamics simulations were performed on the Thigh, Genu, and Calf-1 domain of αIIb integrin (residues P452 to R743) that constitute the “knee joint.” Ablation of the C490-C545 disulfide bond results in clearly different conformational distributions of the domains, implying a major effect of the bond on the dynamics of the knee joint (Figure 5B). Force network analysis indicates that ablation of the C490-C545 disulfide bond results in propagation of subtle mechanical forces throughout the protein structure (Figure 5C), whereas force calculations suggest that the closed form of the integrin is disfavored when the C490-C545 disulfide is missing, reducing cycling efficiency of the integrin (Figure 5D; supplemental Figure 8; supplemental Table 2). These studies imply that lack of the C490-C545 disulfide bond changes the forces required for bending at the αIIb knee joint.
Humans express 18 integrin α subunits. An important question is whether this alternate αIIb integrin form and function are found in other α subunits.
The αIIb C490, C545 cysteine pair is conserved across all 18 integrin α subunits, and the bond in the αV and α2 subunits is similarly missing
Alignment of the amino acid residues of the 18 human integrin α subunit Thigh domains indicates that the αIIb C490-C545 disulfide is conserved across all α subunits (supplemental Figure 9A). This finding suggested that the alternate αIIb integrin form may also be conserved. This was tested by measuring the redox state of the equivalent disulfide bond in αV (C478-C535) of αVβ3 in the U251 human glioma cell line and α2 (C651-C708) of α2β1 in human umbilical vein endothelial cells. The αV bond is missing in ∼1 in 3 molecules and the α2 bond in ∼1 in 5 molecules expressed by the cells (supplemental Figure 9B), implying that the alternate α subunit form is conserved.
Discussion
Disulfide bonds are the covalent links between pairs of cysteine amino acids and are abundant in secreted and membrane proteins.41 These bonds were traditionally believed to be fully formed and inert in the mature protein, although recent findings indicate that this is not the case for some secreted soluble human proteins. Thirteen fibrinogen disulfide bonds and 12 α2-macroglobulin disulfides are missing in 10% to 50% and 10% to 70% of the molecules, respectively, in the blood of healthy human donors.27 The functional relevance of the disulfide lability for fibrinogen conversion to fibrin polymer has been studied. Disulfides form upon fibrin polymerization and are important for a stable fibrin matrix. In addition, the disulfide-bonded states of fibrinogen in plasma are influenced by fluid shear forces, indicating that the different covalent states can change in response to an external stimulus. Here we report different disulfide-bonded states of the plasma membrane αIIbβ3 integrin receptor.
Human platelets contain an alternate covalent form of αIIbβ3 integrin that constitutes ∼1 in 3 molecules of the total integrin pool. The disulfide bond that normally links cysteines 490 and 545 in the αIIb subunit is not formed in this species. Human megakaryoblasts and BHK cells transfected with recombinant human integrin produce the same alternate covalent form with the same prevalence, implying that it is predetermined in megakaryocytes and not a consequence of posttranslational changes at the cell surface. In support of this conclusion, the redox state of the C490-C545 disulfide is not influenced by platelet activation, and the bond in recombinant αIIbβ3 integrin is not cleaved by any of the 5 known circulating disulfide-cleaving enzymes or vascular thiol isomerases.30,31 It is of interest that the C490-C545 disulfide bond in the X-ray structure of the extracellular region of resting αIIbβ3 integrin (PDB identifier 3fcs7) has a –RHstaple conformation, which is a disulfide configuration that is naturally strained. Disulfide bonds are classified based on the geometry of the 5 dihedral angles that define the cystine residue,42 and 2 of the 20 different conformations, the –RHstaple and −/+RHhook disulfides, have topological features that stress the disulfides through direct stretching of the sulfur–sulfur bond and neighboring angles.42,43 The energy barrier for formation of the C490-C545 disulfide bond is predicted to be higher than for other αIIb disulfides, which could be a factor that contributes to production of an αII integrin missing the bond.
The alternate covalent form of αIIbβ3 integrin partitions into focal adhesions on the activated platelet surface and in BHK cells expressing a mutated αIIbβ3 integrin with an ablated C490-C545 disulfide bond. The incidence of the alternate form is ∼1 in 3 molecules in the total platelet αIIbβ3 integrin pool, compared with ∼2 in 3 molecules in focal adhesions in activated platelets. This selective partitioning of the alternate αIIbβ3 form on the cell surface suggested that it has differentiating properties. Indeed, the disulfide mutant integrin in BHK cells has extended residency time in focal adhesions due to a reduced rate of clathrin-mediated integrin internalization and recycling. Internalization of integrins is regulated by the clathrin adaptor protein AP2.36 AP2 binds to the cytoplasmic domain of αIIbβ3 and mediates cargo recruitment, maturation, and scission of the endocytic vesicle.37 Inhibition of integrin recycling increases AP2 association with αIIbβ3,44 implying that disassociation of AP2 from αIIbβ3 is necessary for integrin recycling. There is enhanced binding of AP2 for the αIIb subunit when the C490-C545 bond is missing, which is consistent with the restricted integrin recycling.
Conformational differences in αIIbβ3 integrin with an intact vs missing C490-C545 bond were investigated by using molecular dynamics simulations. Changes in bending at the αIIb knee joint were explored. Ablation of the C490-C545 bond results in propagation of subtle mechanical forces throughout the Thigh, Genu, and Calf-1 domains, and force calculations indicate a higher energy barrier for cycling of the integrin between bent and partially open forms when the C490-C545 disulfide is missing. In other words, the integrin form with a missing C490-C545 disulfide bond requires higher forces to transition from bent to open conformational states. This observation is consistent with the reduced affinity for fibrinogen and activation by manganese ions of the alternate covalent form of αIIb.
Integrin subsets in focal adhesions with extended residency time and states of activation have been reported. In photobleaching experiments, Legerstee et al45 observed that paxillin and vinculin in focal adhesions exist in stably bound, dynamically bound, or mobile pools. Approximately 40% of the focal adhesion proteins were in the stably bound pool, implying a reduced rate of cycling of the associated integrins in this pool. Using super-resolution microscopy techniques and antibodies that bind to different integrin conformations, Spiess et al46 observed nanoclusters of both active and inactive β1 integrins in focal adhesions. The longer lived and inactive integrins in focal adhesions described in these studies share features with the alternate αIIbβ3 covalent form characterized herein. The αIIb C490, C545 cysteine pair is conserved across all 18 human integrin α subunits, and the bond in αV and α2 subunits in cultured cells is similarly missing. The finding that the same disulfide bond in different α subunits on different cells share very similar features suggests that the alternate integrin form and function are conserved across integrins.
Our observation that integrins are produced in different covalent forms that have different functions has broad implications for the many physiological and pathological cellular processes that these receptors participate in.47 An important question is whether there are >2 covalent forms of α subunits; that is, states in addition to those with an intact or missing C490-C545 bond. Notably, the αIIb C56-C65, C473-C484, and C674-C687 disulfide bonds are missing in ∼1 in 5 integrin molecules in platelets from healthy human donors. Future experiments will explore the distribution and function of these other potentially distinct αIIb forms. Our findings also have implications for therapeutic targeting of integrins. If a therapeutic binds preferentially to one or more covalent forms of an αIIbβ3 integrin, then events that change the incidence of the forms would result in a variable drug response. Indeed, there is significant variation in the incidence of the alternate αIIbβ3 covalent form in healthy donors, which ranges from 7% to 45% of the total integrin pool. We are measuring changes in the incidence of the alternate form in patients with cardiovascular diseases to test this hypothesis.
Acknowledgments
M.T.B. acknowledges helpful discussions with Hugo Macdermott-Opeskin and Cassidy Whitefield, and an Australian Government Research Training Program Scholarship and Dean’s Merit Scholarship in Science. A.E.P. acknowledges the NHMRC Clinical Trials Centre at the University of Sydney, as well as Sydney Catalyst for a stipend scholarship and Top Up award, respectively. The authors thank all the blood donors for their kind contributions. This research was funded by the National Health and Medical Research Council of Australia (grants 1110219, 1143400, and 1143398; P.J.H.) and a Senior Researcher Grant from the NSW Cardiovascular Research Capacity Program (P.J.H.).
Authorship
Contribution: P.J.H. conceived the study, designed experiments, and cowrote the manuscript; A.E.P. designed and performed experiments and cowrote the manuscript; and M.T.B., M.L.C., F.P., and J.C. designed and performed experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Philip J. Hogg, The Charles Perkins Centre, Education and Research Hub – D17, The University of Sydney, NSW 2006 Australia; e-mail: phil.hogg@sydney.edu.au.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE49 partner repository with the dataset identifier PXD025907. All other relevant data are available from the corresponding authors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal