

In this issue of Blood, 1 provide experimental analysis followed by thoughtful discussion, including the potential risks of modifying the pathophysiology of sickle cell anemia (SCA) with therapeutic agents that increase oxygen affinity.
Two generations ago, SCA was recognized as the first “molecular disease.”2 This launched decades of research into normal and sickle hemoglobin (HbA and HbS, respectively). These efforts elucidated the chemistry and biophysics of normal hemoglobin oxygen transport, as well as the physiology of oxygen delivery and the pathophysiology of intracellular HbS polymerization. The recent development of drugs for SCA that target these processes requires that hematologists become reacquainted with this “old” knowledge.
The fundamental role of hemoglobin is the transport of oxygen. This process, crafted by evolution, is a remarkably adaptable system with near-complete saturation of hemoglobin with oxygen in the lungs and efficient oxygen delivery to tissues. Differential oxygen loading and unloading is a result of changing Hb-O2 affinity, made possible by a sigmoidal oxygen dissociation curve (ODC), which describes the fractional oxygen saturation of hemoglobin as a function of the partial pressure of oxygen. Unlike myoglobin, the binding of oxygen to hemoglobin exhibits cooperativity, whereby its oxygen affinity increases as more oxygen is bound. With binding of more oxygen molecules to heme, stepwise conformational changes occur in the hemoglobin tetramer that increase Hb-O2 affinity. In the case of hemoglobin binding oxygen, the more you have, the more tightly you hold.
This cooperative binding of oxygen by hemoglobin is accurately described by a 2-state model,3 in which equilibrium exists between 2 hemoglobin states defined by quaternary structure: the R (relaxed) state with high oxygen affinity and the T (tense) state with low oxygen affinity. Hb-O2 affinity is also modulated physiologically by temperature, pH, and 2,3-diphosphoglycerate (DPG), a small metabolite of anaerobic glycolysis that binds preferentially to deoxygenated hemoglobin. In response to changing oxygen requirements of metabolically active tissues, DPG shifts the ODC to the right, enabling easier oxygen unloading.
The ODC is often reported as the p50 value, which is the partial pressure of oxygen at which hemoglobin is half saturated with oxygen. Importantly, this number reflects the average oxygen saturation across all erythrocytes, which vary individually by mean corpuscular hemoglobin concentration (MCHC), fetal hemoglobin (HbF), and amount of DPG. Further, p50 is usually measured in nonphysiological equilibrium conditions of slow deoxygenation over minutes, rather than the physiological conditions of rapid deoxygenation that occurs in tissues over seconds. Consequently, p50 is a simplified parameter that belies much complexity and should be interpreted accordingly.
Oxygen delivery is vital for life, but is also detrimental to sickle erythrocytes. The HbS tetramer (α2βS2) begins to polymerize rapidly when it shifts from the oxygenated R state to the deoxygenated T state. After oxygen is delivered to the tissues, sickle erythrocytes undergo changes in shape that block blood flow, and this vaso-occlusion impedes further oxygen delivery. Theoretically, if the oxygen affinity of HbS were increased, then the R state would be favored, and HbS polymerization would be delayed, which should be of therapeutic benefit. The challenge for drugs that increase oxygen affinity is to shift the oxygen affinity just enough to help delay sickling but not enough to impair oxygen delivery (see figure). The goal is to have enough, but not to hold too much.
This is the intentional design of voxelotor,4 a small molecule that binds covalently to the α-globin chain within the HbS tetramer and helps stabilize the high-affinity R state that does not polymerize. Voxelotor was approved by the US Food and Drug Administration in 2019 after a randomized, multicenter, phase 3 trial demonstrated that 1500 mg once daily improved anemia by >1 g/dL, compared with placebo, and also reduced laboratory markers that reflect hemolysis.5 In that initial report, there was no improvement in rates of vaso-occlusive pain with voxelotor, which was confirmed in a recent long-term follow-up report.6
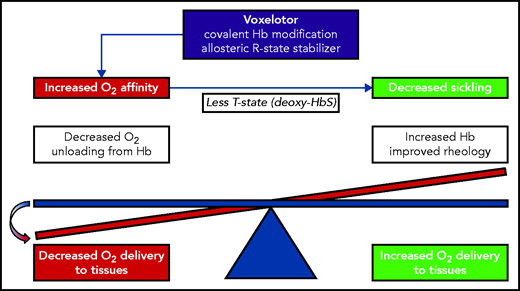
The balance between oxygen affinity of hemoglobin and oxygen delivery to the tissues. In SCA, increasing the oxygen affinity should delay hemoglobin polymerization but at a potential cost of decreasing oxygen delivery. The experimental results of Henry et al indicate that voxelotor treatment tips the scale toward decreased oxygen delivery.
The balance between oxygen affinity of hemoglobin and oxygen delivery to the tissues. In SCA, increasing the oxygen affinity should delay hemoglobin polymerization but at a potential cost of decreasing oxygen delivery. The experimental results of Henry et al indicate that voxelotor treatment tips the scale toward decreased oxygen delivery.
Henry and colleagues present experimental analyses that address this apparent paradox, specifically how inhibition of HbS polymerization could fail to improve clinical symptoms of SCA and especially pain, the hallmark symptom of vaso-occlusion. Using a novel in vitro sickling assay with an automated oxygen gradient that recapitulates rapid physiologic deoxygenation, they demonstrate that voxelotor produces the predicted left shift in the ODC with lowering of p50, which inhibits HbS polymerization. However, this effect comes at the cost of impaired tissue oxygen delivery, unless better blood flow ensues (see figure). This theoretical concern about voxelotor was raised previously, particularly in the setting of cerebrovascular blood flow,7 but to date, few experimental data have been available to guide clinicians.
Henry et al also provide data regarding the importance of HbF and MCHC in SCA, suggesting that these are better therapeutic targets for prevention of HbS polymerization. HbF forms hybrids with HbS (α2βSγ) that inhibit polymerization, but do not cause a left shift in the ODC. Accordingly, HbF induction has proven therapeutic benefit for SCA, mainly through decades of research using hydroxyurea,8 although other agents are currently under investigation. Similarly, lowering the MCHC through improved cellular hydration delays HbS polymerization, again without any predicted effect on oxygen affinity or delivery. To date, no approved therapeutic agents specifically target the MCHC, and previous efforts to reduce cellular dehydration were unsuccessful at reducing vaso-occlusive pain.9
Ultimately, the safety of a drug can be demonstrated only in human clinical trials with careful long-term surveillance. Voxelotor is commercially available and can increase circulating hemoglobin concentration, which improves oxygen-carrying capacity. Whether its net effect in SCA is detrimental by shifting the ODC or beneficial by improving blood flow, remains incompletely defined, but the results of Henry et al should give us pause. Tissue perfusion is challenging to quantify, but measures such as near infrared spectrometry should be studied prospectively, along with serial brain magnetic resonance imaging, to identify parenchymal damage from impaired oxygen delivery. At least for the delicate balance between Hb-O2 affinity and oxygen delivery in SCA, to have and to hold may not always be the best approach.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal