

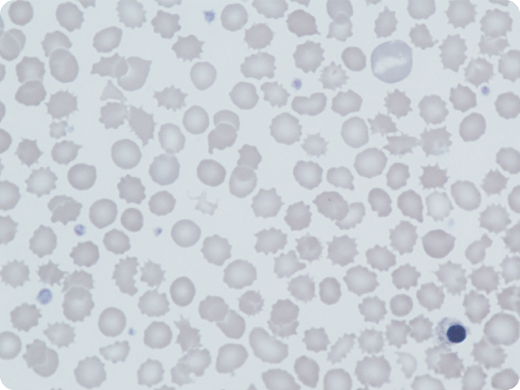
A 5-year-old boy presented with mild neonatal jaundice and hemolytic anemia; his laboratory tests revealed the following: hemoglobin (Hb), 99 g/L; reticulocytes, 19%; mean corpuscular volume (MCV), 71 fL; HbA2, 3.6%; and fetal Hb (HbF), 21%. His parents, from Pakistan, were distantly related; there was no family history of anemia. A blood film showed polychromasia and marked anisopoikilocytosis; in particular, acanthocytes, echinocytes, polychromasia, and occasional nucleated red cells were seen (May-Grünwald-Giemsa stain; original magnification ×100). Erythrocyte pyruvate kinase (PK) activity was low (1.8 IU/g Hb; normal, 13-17 IU/g Hb). Both parents had normal PK activity. A second male sibling had similar hematological findings. Hemolysis, low MCV, and high HbF levels were common features. Sequencing of α-globin, β-globin, and PKLR genes was normal in both brothers but a subsequent red cell gene panel showed that they were both homozygous for a novel Krüppel-like factor 1 (KLF1) variant, c.938G>T; p.Trp313Leu, likely pathogenic by American College of Medical Genetics and Genomics (ACMG) standards and guidelines. Both parents were heterozygous for this variant, which has never previously been described in any populations.
Constitutional KLF1 mutations are suggested by hemolytic anemia with low PK activity, low mean corpuscular Hb/MCV, and persistent high HbF levels. KLF1 is an important transcriptional regulator of erythropoiesis, affecting protein synthesis including globins, heme, cytoskeletal proteins, and enzymes. Morphological features and erythrocyte PK activity in KLF1 mutations simulate PK deficiency. The correct diagnosis is confirmed by gene sequencing.
A 5-year-old boy presented with mild neonatal jaundice and hemolytic anemia; his laboratory tests revealed the following: hemoglobin (Hb), 99 g/L; reticulocytes, 19%; mean corpuscular volume (MCV), 71 fL; HbA2, 3.6%; and fetal Hb (HbF), 21%. His parents, from Pakistan, were distantly related; there was no family history of anemia. A blood film showed polychromasia and marked anisopoikilocytosis; in particular, acanthocytes, echinocytes, polychromasia, and occasional nucleated red cells were seen (May-Grünwald-Giemsa stain; original magnification ×100). Erythrocyte pyruvate kinase (PK) activity was low (1.8 IU/g Hb; normal, 13-17 IU/g Hb). Both parents had normal PK activity. A second male sibling had similar hematological findings. Hemolysis, low MCV, and high HbF levels were common features. Sequencing of α-globin, β-globin, and PKLR genes was normal in both brothers but a subsequent red cell gene panel showed that they were both homozygous for a novel Krüppel-like factor 1 (KLF1) variant, c.938G>T; p.Trp313Leu, likely pathogenic by American College of Medical Genetics and Genomics (ACMG) standards and guidelines. Both parents were heterozygous for this variant, which has never previously been described in any populations.
Constitutional KLF1 mutations are suggested by hemolytic anemia with low PK activity, low mean corpuscular Hb/MCV, and persistent high HbF levels. KLF1 is an important transcriptional regulator of erythropoiesis, affecting protein synthesis including globins, heme, cytoskeletal proteins, and enzymes. Morphological features and erythrocyte PK activity in KLF1 mutations simulate PK deficiency. The correct diagnosis is confirmed by gene sequencing.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal