

In this issue of Blood, report that alterations in the function of TP53 and the cell-cycle regulators CDKN2A/CDKN2B cooperate in Richter transformation (RT) of chronic lymphocytic leukemia (CLL).1 RT is characterized by the development of an aggressive lymphoma from indolent CLL and occurs in 2% to 10% of CLL patients. It represents the most difficult therapeutic challenge in CLL despite the multitude of targeted therapies that are currently available. The estimated overall survival is currently only 3.3 months.2,3 Several factors for the development of RT have been identified such as loss of TP53, NOTCH1 mutations, constitutively active AKT, stereotyped B-cell receptors, and, notably, disruptions of the cell-cycle regulators CDKN2A/B in about 30% of cases.4,5 Because RT is the biggest unmet need for patients with CLL, it is of critical importance to functionally dissect risk factors for RT to provide novel approaches for therapy of RT.
Given these challenges, the work of Chakraborty et al is particularly exciting; the authors show that a cooperative biallelic loss of function of TP53 and the cell-cycle regulators CDKN2A/CDKN2B allows for B-cell receptor (BCR)-dependent proliferation and may provide therapeutic targeting of RT by combining BCR and CDK4/6 inhibitors.
A well-known phenomenon of CLL cells is central to this work. In vitro–cultured CLL cells do not proliferate to a BCR stimulus. Likewise, CLL cells stop proliferating once treated with BCR inhibitors in vivo. The initial studies of the authors explored gene expression data from primary human CLL cells after ex vivo stimulation of the BCR. Here, they identify strong induction of G1 phase cell-cycle regulators, which was counterbalanced by upregulation of the negative regulators CDKN2A and CDKN2B (see figure, panel A). Importantly, these observations from patient CLL cells were recapitulated by the authors in the Eµ-TCL1 mouse model using the autoantigen phosphatidylcholine to provide tonic BCR stimulation. In the murine setting, CLL cells similarly displayed induction of CDKN2A and CDKN2B once stimulated via BCR signaling. This upregulation of the cell-cycle inhibitors CDKN1A, CDKN2A, and CDKN2B upon BCR engagement might mediate control of proliferation in BCR-stimulated CLL cells in the absence of other costimulatory signals. Continued counterbalance in CLL cells may be a defining regulatory roadblock against uncontrolled proliferation and of particular relevance for the indolent characteristics of CLL. Here, the implication that the genetic aberrations with the loss of cycle inhibitors CDKN1A, CDKN2A, and CDKN2B are critical for the transformation from CLL to RT (see figure, panel B). These genes are frequently found to be nonfunctional in patients with RT where ∼50% of patients harboring alterations of TP53, an important transcriptional activator of CDKN1A, and another 30% of patients with deletions involving the CDKN2A locus.4 In an attempt to duplicate the genetic alterations of RT in vivo within the Eµ-TCL1 mouse model of CLL, it has been previously shown that loss of TP53 function induces a high-risk CLL phenotype, but with low rate of progression to RT.6 These findings suggested that additional loss of cell-cycle control may be required for complete transformation to the RT phenotype.
Chakraborty et al have now elegantly addressed the functional consequences of these genetic events occurring in CLL progression to RT. Also, by using the Eµ-TCL1 mouse model for CRISPR/Cas9-based targeting ex vivo, they addressed simultaneous loss of TP53, CDKN2A, and CDKN2B in murine CLL cells derived from Eµ-TCL1 mice. On a technical level, it is noteworthy that the investigators achieved effective targeting in this model by using a 2-step in vivo expansion of Cas9-targeted CLL cells. The key finding of their experimental approach was that the combined targeting of TP53, CDKN2A, and CDKN2B induces a diffuse RT-like morphology with infiltration of large pleomorphic cells with a high proliferative rate. Furthermore, the TP53-, CDKN2A-, and CDKN2B-deficient cells were able to spontaneously proliferate ex vivo, whereas wild-type control cells could not even be maintained ex vivo. These alterations were specific for the combined loss of TP53, CDKN2A, and CDKN2B function, whereas targeting the respective genes individually did not produce increased proliferation ex vivo.
The authors also addressed these findings in an independent BCR specificity setting using a Smith autoantigen reactive variant of the Eµ-TCL1 model for targeting of TP53, CDKN2A, and CDKN2B function. Also, with this alternative BCR specificity, the combined loss of the respective cell cycle inhibitors again revealed an aggressive RT phenotype with significantly reduced overall survival and large cells with the propensity to proliferate ex vivo. Thus, biallelic loss of function of TP53 and the cell-cycle regulators CDKN2A/CDKN2B paves the way for stimulus-induced proliferation in CLL cells in RT.
When altering these regulators of key processes such as cell-cycle control and DNA damage response, one might hypothesize that additional off-target genetic alterations driving proliferation of CRISPR/Cas9-edited cells are necessary. However, whole exome sequencing of targeted cells did not reveal any additional mutations underlying the RT phenotype and the increase in proliferation.
One might expect an elevated load of additional mutations from genomic instability in the context of TP53 loss. However, transformation did not depend on acquisition of additional recurrent mutations. Moreover, the combination of BCR stimulus and unleashed cell-cycle control through loss of regulators CDKN2a/CDKN2B appears to suffice as a breach in the dike toward massively accelerated flood of cellular proliferation in RT.
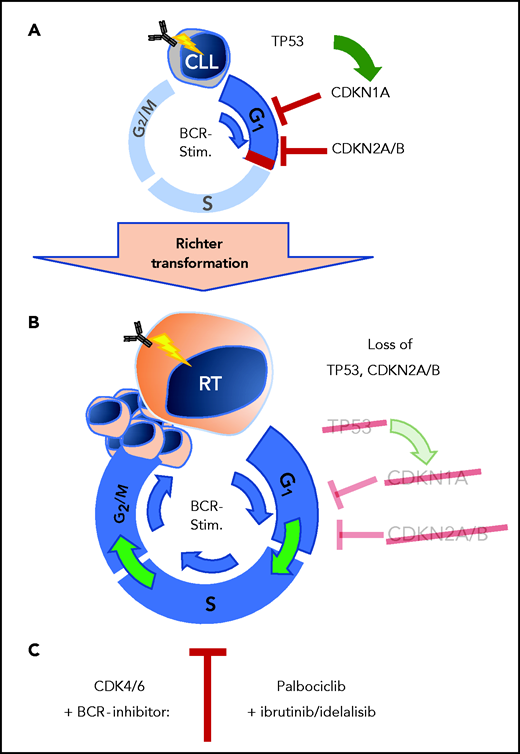
Loss of cell-cycle control in RT from CLL cells. (A) Cell-cycle control by TP53, CDKN1A, CDKN2A, and CDKN2B blocks proliferation in CLL stage B cells after BCR stimulation. (B) Loss of TP53 and CDKN2A/B in RT allows transformed cells to proliferate past inactivated cell-cycle checkpoints. (C) Combined BCR- and CDK4/6-inhibition provides effective therapeutic response in preclinical RT mouse models. BCR-stim., BCR stimulation.
Loss of cell-cycle control in RT from CLL cells. (A) Cell-cycle control by TP53, CDKN1A, CDKN2A, and CDKN2B blocks proliferation in CLL stage B cells after BCR stimulation. (B) Loss of TP53 and CDKN2A/B in RT allows transformed cells to proliferate past inactivated cell-cycle checkpoints. (C) Combined BCR- and CDK4/6-inhibition provides effective therapeutic response in preclinical RT mouse models. BCR-stim., BCR stimulation.
The loss of cell-cycle control by loss of regulator function obviously provides the path for massive acceleration of proliferation underlying RT. The question remained as to which pathway was the predominant culprit driving CLL cells to increased proliferation. To explore that question, the authors compared BCR and TLR signaling pathways by targeting the IgM constant region vs MyD88 respectively. Suppression of BCR signaling by disruption of IgM abrogated proliferation and progression in vivo. Conversely, targeting of the TLR adaptor protein MyD88 did not alter malignant cell growth. The authors conclude that proliferation from biallelic loss of TP53/CDKN2A/2B is dependent on BCR signaling and does not require other costimulatory signals from the leukemic microenvironment.
The merit of these insights is that they provide a functional rationale for the genomic alterations underlying RT. This work also provides a therapeutic strategy toward treatment in the highly resistant and notoriously difficult to target RT. The authors observed BCR inhibitors terminated cell growth in TP53/CDKN2A/B-deficient TCL1-derived cells. Moreover, their work provides a rationale for inhibiting cell-cycle progression via CDK4/6 inhibitors to substitute for the lost control by CDKN2A/B. Using the CDK4/6 inhibitors palbociclib, a well-established compound in breast cancer frontline therapy, they observed inhibition of proliferation of RT cells. When combining palbociclib with BCR-signaling directed compounds ibrutinib, idelalisib, or fostamatinib, a synergistic effect was observed in vitro. Consistently, a significant response to combined BCR plus CDK4/6 targeting was observed in vivo (see figure, panel C). Mice treated with the combination of ibrutinib and palbociclib showed a lower leukemic burden and an increased overall survival. Finally, the authors could replicate their finding based on the TCL1 mouse model by using human xenograft RT cells comparing 2 RT-derived models with either loss of TP53/CDKN2A/B vs maintained integrity of respective loci.7 Here, it is noteworthy that in TP53/CDKN2A/B-deficient RT cells were able to proliferate spontaneously in vitro, whereas RT cells with functional TP53/CDKN2A/B but trisomy 12, KRAS, MED12, and NOTCH2 mutation driving RT did not display this phenomenon. The murine and human model systems of TP53/CDKN2A/B loss display a similar consequence for enhanced and less controlled proliferation, one of the major hallmarks of cancer. Moreover, therapeutic response to combined BCR plus CDK4/6-inhibition consistently showed a synergistic response in the human patient-derived xenograft model.
These results open a multitude of further questions and opportunities for development of clinical approaches. So far, CDK4/6 inhibitors have not been explored in RT, but promising early phase 1 result in mantle cell lymphoma suggest a clear rationale to address ibrutinib in combination with palbociclib in RT as well.7 This study provides a clear clinical rationale. It also provides a new mouse model recapitulating a genetic aberration found in many patients with RT. The development of this new in vivo model of RT in concert with models representing alternative mechanisms of RT pathogenesis will accelerate preclinical exploitation of novel therapeutic concepts.5,9
Conflict-of-interest disclosure: C.P.P. has served as consultant for Abbvie; has received speakers honoraria from Roche, Astellas, and Abbvie; and has received research funding from Gilead and Sanofi.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal